Clinical and genetic features of a Chinese family with ATF6-associated achromatopsia
Zhu Tian, Li Hui, Wei Xing, Wu Shijing, Sun Zixi, Sui Ruifang
Department of Ophthalmology, State Key Laboratory of Complex Severe and Rare Diseases, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100730, China
Corresponding author: Sui Ruifang, Email: hrfsui@163.com
[Abstract] [View PDF in English] [View PDF in Chinese] [Read Full Text]
Objective To identify the clinical characteristics and pathogenic gene of a Chinese Han family with achromatopsia (ACHM).
Methods The method of pedigree investigation was adopted. A Chinese Han ACHM family was recruited in Peking Union Medical College Hospital from July 2010 to July 2019, including five members of two generations. There were two patients and three phenotypically normal individuals. The medical history was obtained and comprehensive ophthalmic examinations were performed, including vision acuity, color vision, color fundus photography, fundus autofluorescence, optical coherence tomography (OCT), visual field, and electroretinogram (ERG). Genomic DNA was extracted from a peripheral blood sample from the patients and family members. The pathogenic variant was screened by whole exome sequencing and was verified by Sanger sequencing and co-segregation analysis. The variant was annotated with the Human Gene Mutation Database and the 1000 Genomes, Exome Aggregation Consortium (ExAC), ClinVar, and Online Mendelian Inheritance in Man (OMIM) databases to detect the single nucleotide polymorphism and whether it had been reported previously. The pathogenicity of the variant was evaluated according to the standards and guidelines of the American College of Medical Genetics and Genomics (ACMG).
Results There was consanguinity between the proband’s parents and this family was consistent with autosomal recessive inheritance. Both male patients presented a reduction of visual acuity accompanied by photophobia and color blindness since childhood. A barely visible foveal light reflex in fundus images and hypofluorescence of foveal areas in FAF images were found. The foveal structure was invisible and disruption of the ellipsoid and interdigitation zones were missiong in OCT images in both patients. Perimetry showed a central scotoma with or without peripheral defects. ERG revealed that scotopic 0.01, 3.0, and 10.0 stimulation responses were normal, oscillatory potential amplitudes were
reduced, and photopic 3.0 stimulation responses or 30Hz flicker responses were irrecordable. No sign of progression was found
during a 9-year follow-up. It was confirmed that both patients carried a novel homozygous disease-causing variant c.947insA (p.Asn316Lysfs*46) in the ATF6 gene. Their mother had the heterozygous variant. The unaffected brother did not carry the variant. This family was consistent with co-segregation. This variant was labeled as pathogenic according to the ACMG classification.
Conclusions A novel variant c.947insA (p.Asn316Lysfs*46) in the ATF6 gene is the pathogenic mutation of this family with achromatopsia. To our knowledge, this is the first time that this variant has been reported.
[Key words] Achromatopsia; Pedigree; Genetic testing; Whole exome sequencing; ATF6 gene
Fund program: National Natural Science Foundation of China (81873687)
DOI: 10.3760/cma.j.cn115989-20210909-00506
Achromatopsia (ACHM), also known as rod monochromatism, is an autosomal recessive inherited disorder of cone cell dysfunction, with low vision, photophobia, nystagmus, and dyschromatopsia since childhood as major manifestations. Six genes have been reported to be associated with ACHM: CNGB3 (MIM 605080), CNGA3 (MIM
600053), GNAT2 (MIM 139340), PDE6C (MIM 600827), PDE6H (MIM601190), and ATF6 (MIM605537) [1-6]. Among them, the ATF6 gene was recently reported to be associated with ACHM. To date, 21 pathogenic variants have been found in the ATF6 gene, of which 18 are from European and American patients and only 3 are from Chinese patients [6-13]. The reported pathogenic variations of the ATF6 gene in Chinese patients were determined by large-sample gene screening, but detailed information on clinical phenotype is lacking [10]. In the present study, a Chinese Han family with ACHM caused by a novel pathogenic variation of the ATF6 gene was identified by whole exome sequencing (WES). The clinical phenotype of two patients was described in detail and followed up for a significant time period, and the characteristics of its natural course were summarized.
1 Data and Methods
1.1 General data
A pedigree investigation was performed. A Chinese Han family with ACHM treated in the Ophthalmology Department of Peking Union Medical College Hospital in China from July 2010 to July 2019 was recruited, including five members of two generations. There were two patients and three phenotypically normal subjects. This study was conducted in accordance with the Declaration of Helsinki and the study protocol had been reviewed and approved by the Ethics Committee of Peking Union Medical College Hospital, China Academy of Medical Sciences (Approval No.: JS-2059). The patients’ legal guardians were informed of the study objectives and voluntarily signed the informed consent form.
1.2 Methods
1.2.1 Clinical examinations The patients’ history of eye diseases, family history of eye diseases, and marriage and childbearing history of their parents were recorded in detail. The visual acuity of the subjects was examined using a standard logarithmic visual acuity chart. The subjects’ color vision, fundus, retinal structure, visual field, and retinal function were detected by a color vision test chart and D-15 color disc test, ultra-wide-angle color fundus photography and fundus autofluorescence (FAF, Optos, Dunfermline, Scotland KY11 8GR, UK), optical coherence tomography (OCT, Heidelberg, Germany), perimeter (Octopus, Switzerland), and electroretinography (ERG, Roland Consult, Germany), respectively. Regarding ERG, the examination and analysis were conducted according to the standards published by the International Society for Clinical Electrophysiology of Vision. ERG-Jet contact lens electrodes were used in the two patients.
1.2.2 Molecular genetic testing Peripheral venous blood (5 mL) was drawn from both patients and their relatives (mother and full brothers) (blood samples were not collected from the father for particular reasons). Whole genomic DNA was extracted using the QIAamp DNA Blood Midi Kit (QIAGEN, Hilden, Germany). The coding regions of all genes were subjected to WES, and the sequencing data were compared with the University of California Santa Cruz (UCSC) human genome reference sequence hg19 (GRCh37). The variants were annotated with the Human Gene Mutation Database and the 1000 Genomes, Exome Aggregation Consortium (ExAC), ClinVar, and Online Mendelian Inheritance in Man (OMIM) databases to detect the single nucleotide polymorphism and whether this had been reported previously. The pathogenicity of the variation was evaluated according to the recommended guidelines of the American College of Medical Genetics and Genomics (ACMG) [14]. Suspected pathogenic variations were verified by Sanger sequencing and co-segregation analysis.
2 Results
2.1 Clinical characteristics
The proband’s parents were consanguineous, with normal phenotypes, and some of the offspring were affected. The genetic characteristics of the family conformed to autosomal recessive inheritance (Figure 1).
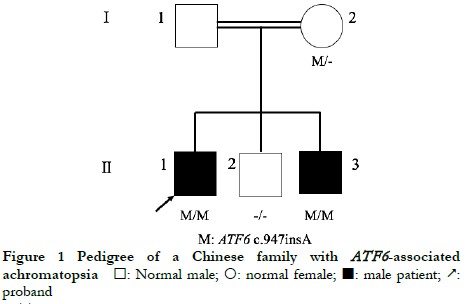
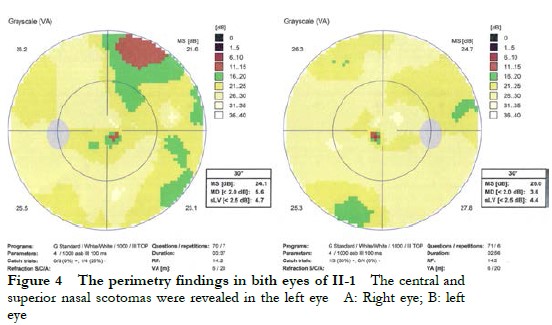
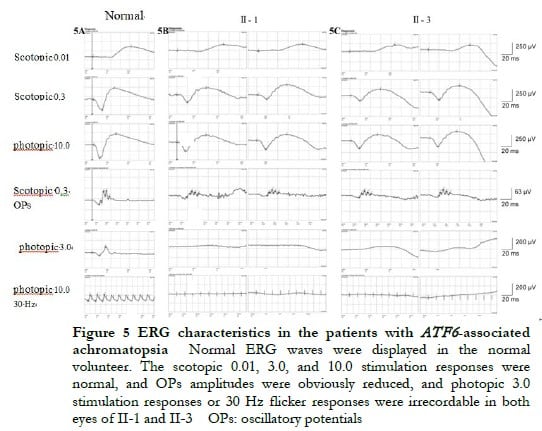
The proband II-1, male, visited a doctor for the first time aged 7 years, having had poor vision and photophobia since childhood. The visual acuity was +2.00 DS + 2.25 DC ´ 110° = 0.1 (right eye) and +1.50 DS + 2.25 DC ´ 75° = 0.15 (left eye) at the first visit, and −0.25 DS + 3.25 DC ´ 112° = 0.12 (right eye) and 0.00 DS + 3.00 DC ´ 74° = 0.15 (left eye) at the last visit when aged 16 years. The proband’s younger brother II-3 visited a doctor for the first time when aged 3 years, and had photophobia and poor vision. This patient could not cooperate in the visual acuity examination, and had nystagmus of both eyes. The visual acuity was −2.75 DS + 4.25 DC ´ 100° = 0.1 (right eye) and −1.00 DS + 3.50 DC ´ 75° = 0.12 (left eye) at the last visit when aged 12 years, and neither eye displayed nystagmus. At the last follow-up, D-15 color disc tests were performed several times on patients II-1 and II-3, and the results showed poor repeatability, but they both showed abnormal color vision (red, green, and blue). The examination results at the last visit are shown in Figure 2. A barely visible foveal reflex was found in the fundus examination, hypofluorescence was observed in the macular fovea of both eyes in FAF, and there was no obvious macular fovea of both eyes with the absence of the ellipsoid and interdigitation zones in the corresponding areas by OCT (Figure 3). The visual field test of proband II-1 showed a small central scotoma in both eyes and a supranasal scotoma in the left eye (Figure 4). ERG results of patients II-1 and II-3 showed that the scotopic 0.01, 3.0, and 10.0 response waveforms were generally normal, the amplitude of the oscillatory potential reduced, and no photopic 3.0 or 30 Hz flicker responses were recorded (Figure 5). During the follow-up period, the clinical manifestations and auxiliary examination results of patients II-1 and II-3 revealed no significant disease progression. The clinical examination results of the proband’s mother I-2 and younger brother II-2 displayed no obvious abnormalities. The proband’s father I-1 underwent visual acuity and fundus examinations in a local hospital, and the results showed no obvious abnormalities.


2.2 Molecular genetic analysis
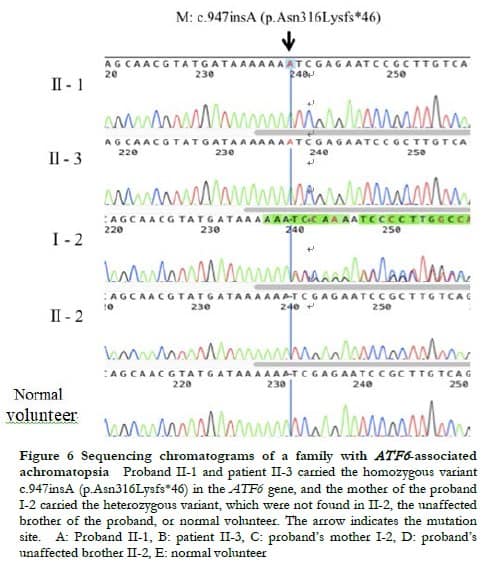
WES showed that both proband II-1 and patient II-3 carried the homozygous variant c.947insA of the ATF6 gene (NM_007348.3), the proband’s mother I-2 carried the heterozygous variant, and the proband’s brother II-2 did not carry the variant. The genetic test results of the patients and their relatives were consistent with familial co-segregation (Figure 6). This variant was not recorded in the control database, thus it was a novel frameshift variation located at chr1:161789454. The variant c.947insA altered the reading frame, causing premature termination of protein translation at amino acid position 362, that is, p.Asn316Lysfs*46. This variant was labeled as pathogenic according to the ACMG classification. Combined with the history of consanguineous marriage of their parents, the variant c.947insA in the ATF6 gene was identified as the pathogenic variant of the two patients.
3 Discussion
Low vision, dyschromatopsia, and photophobia were observed since childhood in the two patients included in this study, suggesting cone system involvement. Cone system involvement includes progressive and static diseases.. ACHM is a common static disease. The fundus manifestations of the two patients showed no obvious abnormalities, while disruption of the ellipsoid and interdigitation zones and macular fovea hypoplasia were observed by OCT, which conformed to the characteristic manifestations of patients with ATF6 gene variation [6]. A considerable reduction was found in the selective cone response by ERG, which is an auxiliary examination with clinical diagnostic significance. For example, the patient II-3 visited a doctor for the first time when aged 3 years, and could thus not cooperate with the visual acuity examination, and OCT could not be performed due to obvious nystagmus. ERG was performed after the patient took sedatives, and typical ERG manifestations were observed, thus the patient was diagnosed with ACHM. During the 9-year follow-up, neither of the two patients in this family displayed any obvious disease progression.
The globally prevalence of ACHM in the population is approximately 1/30,000 [15-16], and 70%–80% of ACHM is caused by CNGA3 or CNGB3 gene variations [17-19]. The CNGA3 gene is a common pathogenic gene in ACHM patients in China and the Middle East, and approximately 60% of such ACHM cases are attributed to CNGA3 gene mutations [20]. The CNGB3 gene is a common pathogenic gene in ACHM patients in Europe and the United States, and its mutations lead to approximately 50% of such ACHM cases [21]. ACHM caused by GNAT2 gene variations accounts for approximately 2% of all ACHM cases [22], while the PDE6H and PDE6C gene variations lead to <2% of ACHM [4]. In 2015, Kohl et al. [6] first reported that the ATF6 gene is associated with ACHM. In the present study, whole genomic DNA was extracted from the blood samples of the proband II-1, the proband’s younger brother II-3, the proband’s mother I-2, and the proband’s unaffected younger brother II-2 for genetic testing at the first visit of the proband in 2010. The pathogenic genes of ACHM such as CNGB3, CNGA3, and GNAT2 were excluded. In 2019, the blood samples were collected again from the above members for WES at the re-examination of the two patients. A novel homozygous variant (c.947insA) of the ATF6 gene was found, which was verified by Sanger sequencing in combination with clinical manifestations to be consistent with familial co-segregation, thus they were diagnosed with ATF6 gene-related ACHM. Due to particular reasons, a blood sample was not collected from the proband’s father I-1 for Sanger sequencing. Despite this, the WES data of the proband’s mother I-2, proband II-1, and proband’s younger brother II-3 showed that the ATF6 gene on chromosome 1 had 17 variants in the proband’s mother and 15 variants in the proband and the younger brother. Moreover, the variants near the ATF6 gene in the proband and the younger brother were heterozygous, thus the occurrence of maternal uniparental disomy (UPD) was excluded. In addition, the probability of UPD occurring on any chromosome is approximately 1/3,500, and the probability of UPD occurring twice in the same family is even less. Combined with the family history of the parents’ consanguineous marriage in the family, this homozygous variant was considered to be consistent with Mendelian autosomal recessive inheritance.
Located on chromosome 1q23.3, the ATF6 gene contains 16 exons. Only 21 variants have been reported to be associated with ACHM to date, most of which are reported in foreign patients (Figure 7) [6- 9, 11 -13]. Sun et al. [10] performed genetic testing by WES on a large sample of patients with retinal degeneration in China, and found that three novel ATF6 gene variations caused the disease in two out of 119 patients with definite pathogenic genes. In combination with the findings of the present study, four ATF6 gene variations (all novel variations) have been reported to be associated with ACHM in China. Among them, one homozygous splicing variant (c.1434-1G>A) in the study of Sun et al. [10] and one homozygous insertion mutation (c.947insA) in the present study led to two cases of ACHM, and two missense mutations (c.1127G>A|c.1456T>G) resulted in one case of cone-rod dystrophy. Due to a small sample size, the relationship between genotype and phenotype has not been clarified.

As one endoplasmic reticulum stress (ERS)-regulated transmembrane transcription factor [23], glycosylated endoplasmic reticulum (ER) transmembrane protein type 2 is encoded by the ATF6 gene, contains 670 amino acids, is expressed in all retinal layers [7], and is associated with macular fovea development and cone cell function. This protein is composed of a tubular ERS-sensing domain connected with a cytoplasmic basic leucine zipper domain across the ER membrane, and is an important regulator in the unfolded protein response (UPR). The UPR is an intracellular signaling mechanism that inhibits ERS and ensures ER homeostasis. After ERS occurs, ATF6 monomer protein will translocate from the ER to the nucleus via the Golgi apparatus to up-regulate the expression of target genes, including ER chaperone protein BiP/GRP78, and inhibits ERS by mediating the UPR, thus restoring cellular homeostasis. ATF6 gene variation leads to a reduction of protein transcription activity, which may lead to ERS disorders and increased apoptosis during retinal development, thus affecting macular fovea development and resulting in ACHM [12-13, 24]. In the present study, the homozygous variant c.947insA was found to have an extra nucleotide A at position 947 of the coding region, which caused frameshift mutation and premature termination of protein translation, ultimately producing a shorter protein product with only 362 amino acids.
In recent years, rapid progress has been made in gene therapy. For example, clinical trials of gene therapy have been conducted for ACHM caused by CNGA3 and CNGB3 gene variations (http://clinicaltrials.gov/). No effective treatment is available for ATF6 gene-related ACHM. Glycerol phenylbutyrate (PBA) has been approved by the American Food and Drug Administration for the treatment of urea cycle disorders, which can effectively reduce ERS that is associated with the ATF6 gene. Therefore, Stephen Tsang’s research team applied for the use of PBA for the treatment of ATF6 gene-related ACHM, but patient recruitment has not been started in this clinical trial (http://clinicaltrials.gov/). In the future, the pathogenesis and treatment of ATF6 gene-related ACHM can be studied by establishing animal models of ATF6 gene variations or inducing differentiation of patient-derived somatic cells into pluripotent stem cells and further into retinal organoids as disease models. At present, symptomatic treatment can be temporarily adopted for ATF6 gene-related ACHM. Patients with photophobia should avoid direct sunlight, and those with ametropia are advised to receive optical services.
In conclusion, the clinical characteristics of a Chinese family with ATF6 gene-related ACHM were described in detail in this study for the first time, and the natural course of ATF6 gene-related ACHM was revealed by a 9-year follow-up period. The findings provide a clinical basis for prognostic evaluation and selection of treatment timing for such patients in the future. However, only one family with such a rare disease was collected in this study, so more family data are needed to verify the conclusion. In addition, a novel variation of the ATF6 gene was identified in this study, which expands the spectrum of variations of the ATF6 gene and provides a basis for further understanding the pathogenesis of this disease.
Conflict of Interest None declared.
Author Contribution Statement Zhu Tian: Direct participation in topic selection, test design, implementation of study, data collection, analysis and interpretation, and drafting; Li Hui, Wei Xing, Wu Shijing, Sun Zixi: data collection and interpretation; Sui Ruifang: Direct participation in topic selection, test preparation and design, data collection, analysis and interpretation, review of informative content of paper, revision of intellectual content, and finalizing
References
[1] Kohl S, Marx T, Giddings I, et al. Total colourblindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cGMP-gated cation channel[J]. Nat Genet, 1998, 19(3):257-259. DOI: 10.1038/935.
[2] Sundin OH, Yang JM, Li Y, et al. Genetic basis of total colourblindness among the Pingelapese islanders[J]. Nat Genet, 2000, 25(3):289-293. DOI: 10.1038/77162.
[3] Kohl S, Baumann B, Rosenberg T, et al. Mutations in the cone photoreceptor G-protein alpha-subunit gene GNAT2 in patients with achromatopsia[J]. Am J Hum Genet, 2002, 71(2):422-425. DOI: 10.1086/341835.
[4] Thiadens AA, den Hollander AI, Roosing S, et al. Homozygosity mapping reveals PDE6C mutations in patients with early-onset cone photoreceptor disorders[J]. Am J Hum Genet, 2009, 85(2):240-247. DOI: 10.1016/j.ajhg.2009.06.016.
[5] Kohl S, Coppieters F, Meire F, et al. A nonsense mutation in PDE6H causes autosomal-recessive incomplete achromatopsia[J]. Am J Hum Genet, 2012, 91(3):527-532. DOI: 10.1016/j.ajhg.2012.07.006.
[6] Kohl S, Zobor D, Chiang WC, et al. Mutations in the unfolded protein response regulator ATF6 cause the cone dysfunction disorder achromatopsia[J]. Nat Genet, 2015, 47(7):757-765. DOI: 10.1038/ng.3319.
[7] Ansar M, Santos-Cortez RL, Saqib MA, et al. Mutation of ATF6 causes autosomal recessive achromatopsia[J]. Hum Genet, 2015, 134(9):941-950. DOI: 10.1007/s00439-015-1571-4.
[8] Lee EJ, Chiang WJ, Kroeger H, et al. Multiexon deletion alleles of ATF6 linked to achromatopsia[J/OL]. JCI Insight, 2020, 5(7):e136041[2022-03-15]. https://pubmed.ncbi.nlm.nih.gov/32271167/. DOI: 10.1172/jci.insight.136041.
[9] Mastey RR, Georgiou M, Langlo CS, et al. Characterization of retinal structure in ATF6-associated achromatopsia[J]. Invest Ophthalmol Vis Sci, 2019, 60(7):2631-2640. DOI: 10.1167/iovs.19-27047.
[10] Sun W, Li S, Xiao X, et al. Genotypes and phenotypes of genes associated with achromatopsia: a reference for clinical genetic testing[J]. Mol Vis, 2020, 26:588-602.
[11] Ritter M, Arno G, Ba-Abbad R, et al. Macular maldevelopment in ATF6-mediated retinal dysfunction[J]. Ophthalmic Genet, 2019, 40(6):564-569. DOI: 10.1080/13816810.2019.1706749.
[12] Chiang WJ, Kroeger H, Chea L, et al. Pathomechanisms of ATF6-associated cone photoreceptor diseases[J]. Adv Exp Med Biol, 2019, 1185:305-310. DOI: 10.1007/978-3-030-27378-1_50.
[13] Skorczyk-Werner A, Chiang WC, Wawrocka A, et al. Autosomal recessive cone-rod dystrophy can be caused by mutations in the ATF6 gene[J]. Eur J Hum Genet, 2017, 25(11):1210-1216. DOI: 10.1038/ejhg.2017.131.
[14] Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med, 2015, 17(5):405-424. DOI: 10.1038/gim.2015.30.
[15] Remmer MH, Rastogi N, Ranka MP, et al. Achromatopsia: a review[J]. Curr Opin Ophthalmol, 2015, 26(5):333-340. DOI: 10.1097/ICU.0000000000000189.
[16] Hirji N, Aboshiha J, Georgiou M, et al. Achromatopsia: clinical features, molecular genetics, animal models and therapeutic options[J]. Ophthalmic Genet, 2018, 39(2):149-157. DOI: 10.1080/13816810.2017.1418389.
[17] Li S, Huang L, Xiao X, et al. Identification of CNGA3 mutations in 46 families: common cause of achromatopsia and cone-rod dystrophies in Chinese patients[J]. JAMA Ophthalmol, 2014, 132(9):1076-1083. DOI: 10.1001/jamaophthalmol.2014.1032.
[18] Zelinger L, Cideciyan AV, Kohl S, et al. Genetics and disease expression in the CNGA3 form of achromatopsia: steps on the path to gene therapy[J]. Ophthalmology, 2015, 122(5):997-1007. DOI: 10.1016/j.ophtha.2014.11.025.
[19] Ding XQ, Harry CS, Umino Y, et al. Impaired cone function and cone degeneration resulting from CNGB3 deficiency: down-regulation of CNGA3 biosynthesis as a potential mechanism[J]. Hum Mol Genet, 2009, 18(24):4770-4780. DOI: 10.1093/hmg/ddp440.
[20] Liang XF, Sui RF, Dong FT. Advances in genetic study of achromatopsia[J]. Chin J Exp Ophthalmol, 2015, 33(8):764-767. DOI: 10.3760/cma.j.issn.2095-0160.2015.08.020.
[21] Michalakis S, Schön C, Becirovic E, et al. Gene therapy for achromatopsia[J/OL]. 2017, 19(3):e2944[2022-03-15]. https://pubmed.ncbi.nlm.nih.gov/28095637/. DOI: 10.1002/jgm.2944.
[22] Moskowitz A, Hansen RM, Akula JD, et al. Rod and rod-driven function in achromatopsia and blue cone monochromatism[J]. Invest Ophthalmol Vis Sci, 2009, 50(2):950-958. DOI: 10.1167/iovs.08-2544.
[23] Shen J, Chen X, Hendershot L, et al. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals[J]. Dev Cell, 2002, 3(1):99-111. DOI: 10.1016/s1534-5807(02)00203-4.
[24] Chiang WC, Chan P, Wissinger B, et al. Achromatopsia mutations target sequential steps of ATF6 activation[J]. Proc Natl Acad Sci U S A, 2017, 114(2):400-405. DOI: 10.1073/pnas.1606387114.