·Clinical research·
Genetic analysis of a Chinese family with a cataract-microcornea syndrome
Zhang Daren1, Lu Lan2, Zeng Jie3, Li Danli4, Wang Yun4, Wang Xizhen4, Huang Li2, Fan Ning4, Liu Xuyang1,5
1Xiamen Eye Center of Xiamen University, Xiamen 361000, China; 2Department of Ophthalmology, Fujian Medical University Union Hospital, Department of Ophthalmology & Optometry, Fujian Medical University, Fuzhou 350001, China; 3Department of Ophthalmology, The 900 Hospital of Joint Logistics Troop of People’s, Liberation Army, Fuzhou 350001, China; 4Shenzhen Eye Hospital, School of Optometry, Shenzhen University, Shenzhen Eye Hospital Affiliated to Jinan University, Shenzhen 518000, China; 5Department of Ophthalmology, Shenzhen People’s Hospital, Shenzhen 514080, China
Corresponding author: Liu Xuyang, Email: xliu1213@126.com
[Abstract] [View PDF in English] [View PDF in Chinese] [Read Full Text]
Objective To analyze the clinical and molecular genetic characteristics of a Chinese family with a congenital cataract-microcornea syndrome.
Methods: The method of pedigree investigation was used. A Chinese Han family with a congenital cataract-microcornea syndrome was recruited at the Xiamen Eye Center of Xiamen University. All family members received a detailed ophthalmologic examination including best-corrected visual acuity, intraocular pressure measurement using a handheld applanation tonometer, slit lamp biomicroscopy, color fundus photography, B-scan ultrasonography, corneal diameter, anterior segment optical coherence tomography, ultrasound biomicroscopy, corneal endoscopy, and corneal topography. Genomic DNA was extracted from peripheral venous blood collected from some patients and unaffected family members. Targeted high-throughput DNA sequencing was performed on the proband. The sequencing chip contained 188 known pathogenic genes related to lens abnormalities. Suspected pathogenic genes were verified by Sanger sequencing in phenotypically normal family members to identify co-segregation and the disease-causing gene. Bioinformatics analysis was performed to analyze the pathogenicity of variants in this family by REVEL. Conserved protein domains were analyzed by InterPro. Physicochemical properties of the mutant protein were analyzed using ProtParam. The deleteriousness of the protein was predicted by PolyPhen-2. Homology analysis of variants in the pathogenic gene used the NCBI website to compare conservation among various species. This study followed the Declaration of Helsinki. The study protocol was approved by the Ethics Committee of Xiamen Eye Center of Xiamen University (No. XMYKZX-LW-2009-003). Written informed consent was obtained from each subject prior to entering the study cohort.
Results There were 39 members of four generations in this family, including 11 patients with an autosomal dominant inheritance pattern. Clinical features of the patients included congenital cataract and microcornea. No obvious abnormality was found using ophthalmic and general examinations. A heterozygous mutation, c.61C>T, in the CRYAA gene was found, resulting in mutation of the amino acids from arginine to tryptophan (p.Arg21Trp) at position 21, consistent with co-segregation. The number of cationic clusters in the mutant protein decreased, and the hydrophilicity and stability were reduced. The variant was predicted to be a deleterious mutation and was highly conserved in multiple species.
Conclusions A novel heterozygous mutation, c.61C>T p.Arg21Trp, in the CRYAA gene was thought to be the causal gene of this family. It is the first time this variant has been reported in China.
[Keywords] Cataract-microcornea syndrome; Pedigree; Genetic testing; Microcornea; Mutation; CRYAA gene
Fund program: Funding was from The National Natural Science Foundation of China (81770924); the Fujian Medical Innovation Project (2017-CX-18); the Basic Research Project of Science and Technology Innovation Committee of Shenzhen (JCYJ20210324125614039); and the Science and Technology Plan Project in Medical and Health of Xiamen (3502Z20194066, 3502Z20194069)
DOI: 10.3760/cma.j.cn115989-20200224-00099
Congenital cataracts account for 10%–38% of blinding eye diseases in children, of which at least 30% are caused by genetic factors [1-2]. Hereditary cataracts are most commonly inherited in an autosomal dominant manner and are phenotypically and genotypically heterogeneous, showing considerable inter- and intrafamilial variabilities [3]. In autosomal dominant congenital cataracts, there is a special phenotype, congenital cataract-microcornea syndrome (CCMC), which accounts for 12%–18% of the disease [4]. CCMC is characterized by congenital cataracts with microcorneas without other systemic disorders or developmental abnormalities, which may be associated with secondary damage to lens dysplasia or mutations in certain growth or transcription factors [5]. To date, more than 100 genes have been reported to be associated with congenital cataracts [5], of which at least eight genes are also associated with corneal dysplasia, including the alpha-A component of alpha-crystallin (CRYAA, MIM 123580), β-crystallin-A4 (CRYBA4, MIM 123631), β-crystallin-B1 (CRYBB1, MIM 600929), β-crystallin-B2 (CRYBB2, MIM). 123620), γ-crystallin-C (CRYGC, MIM 123680), γ-crystallin-D (CRYGD, MIM 123690), α8 connexin (GJA8, MIM 600897), and v-maf avian aponeurosarcoma oncogene homologs (MAF) [4, 6-14]. In this study, the clinical phenotype and molecular genetic characteristics of a four-generation autosomal dominant CCMC family were analyzed to determine the pathogenic genes in the family.
1 Information and methods
1.1 General Information
Using the family investigation method, in July 2019, data from a Han family of Fujian Youxi CCMC was collected from the Department of Ophthalmology, Xiamen Eye Center Affiliated to Xiamen University and the Department of Ophthalmology of Union Hospital Affiliated to Fujian Medical University (Fuzhou, China), with a total of 39 people in four generations, including 11 patients. This study followed the Declaration of Helsinki, and the research protocol was approved by the Medical Ethics Committee of Xiamen Eye Center Affiliated to Xiamen University (approval number: XMYKZX-LW-2009-003), and all subjects were aware of the purpose of this study and voluntarily signed an informed consent form.
1.2 Methods
1.2.1 Medical history collection The 10 members of the family who could be contacted provided basic information to the Ophthalmology Department of Youxi County Hospital of Traditional Chinese Medicine, including name, sex, date of birth, relationship with the proband, place of residence, contact information, previous eye health, general health status, and eye and general condition of relatives of the relevant members including: personal history, previous vision history, whether there were systemic diseases, and histories of eye or systemic surgery or trauma. The follow-up examination of the proband and patients in the family were completed at Xiamen Eye Center Affiliated to Xiamen University, the Department of Ophthalmology, Union Hospital Affiliated to Fujian Medical University, and the Department of Ophthalmology of Youxi County Hospital of Traditional Chinese Medicine.
1.2.2 Eye examination Detailed eye examinations of family members included uncorrected visual acuity (UCVA), best-corrected visual acuity (BCVA), intraocular pressure (SW-500 handheld rebound tonometer; Tianjin Sovi Electronic Technology Co., Ltd, Tianjin, China), slit lamp microscope (SL990N; CSO, Scandicci, Italy), color fundus camera (VISVCAM 524; Carl Zeiss, Jena, Germany), ocular B-type ultrasound (Quantel Medical, Clermont-Ferrand, France), corneal diameter measurement, optical coherence tomography (SS-1000; Tomey, Tokyo, Japan), ultrasound biomicroscopy (Quantel Medical), corneal endothelioscopy (SP-1P; Topcon, Tokyo, Japan), and corneal topography (Pentacam AXL; Oculus, Irvine, CA, USA).
1.2.3 Genomic DNA extraction Genomic DNA extraction involved a total of 2 ml of peripheral venous blood of the subject being collected in an ethylene diamine tetraacetic acid (EDTA) anticoagulant tube. A DNA extraction kit (Oiagen, Hilden, Germany) was used to extract genomic DNA from 200 μl of peripheral blood, and the extracted genomic DNA was quantified and tested for purity using a ultraviolet spectrophotometer(Nanjing Wuyi Technology Co. , Ltd, Nanjing, China).
1.2.4 Gene mutation detection The lens abnormal gene detection chip panel-V3 (containing 188 known pathogenic genes related to lens developmental disorders) was obtained from Beijing MyGenostics Medical Laboratory (Beijing, China) and used to screen for mutations in possible pathogenic genes of the proband. Based on next-generation sequencing technology, genes in the target region were captured and sequenced with an average sequencing depth of 300× per sample. A total of 1–5 μg of DNA and fragments were extracted by enzyme digestion, end repair involved adding “A” to the 3′ end, and then adding a linker to obtain a fragment of 350–400 bp, using the liquid phase capture kit developed by MyGenostics Medical Laboratory. Hybridization, elution, and library amplification were according to standard methods. High-throughput sequencing used a NextSeq 500 sequencer (Illumina, San Diego, CA, USA). The sequencing data were analyzed and filtered, and the screening conditions involved (1) missense, shear, nonsense, synonym, promoter, and read-through mutations; (2) gene mutations in 1,000 genomes, and The Single Nucleotide Polymorphism database (dbSNP) and HapMap databases (www.ncbi.nlm.nih.gov) with a frequency less than 0.01; and (3) gene mutations with homozygosity greater than 10 and heterozygous phases greater than 10. The suspected pathogenic genes and mutations detected by screening were sequenced and verified by Sanger sequencing in other family members to determine whether they were co-isolated from the disease phenotype.
1.2.5 Bioinformatics analysis and variant pathogenicity analysis Bioinformatics analysis and variant pathogenicity analysis involved the allele frequency of variant sites in the normal population retrieved through the dbSNP database. The protein function prediction software, REVEL, was used to perform bioinformatics analysis on the pathogenic genes and their mutations to evaluate the pathogenicity of the variants. InterPro (http://www.ebi.ac.uk/interpro/) was used to analyze protein-conserved domains. The ProtParam tool (http://www.expasy.org/ tools/protparam.html) was used to analyze the physicochemical properties of mutant proteins. The effect of mutations on protein function was predicted using PolyPhen-2 online software (http://genetics.bwh.harvard.edu/pph2/index.shtml). The mutation sites of disease-causing genes were analyzed for homology using the NCBI website to compare their conservation with different species.
2 Results
2.1 Clinical phenotype analysis
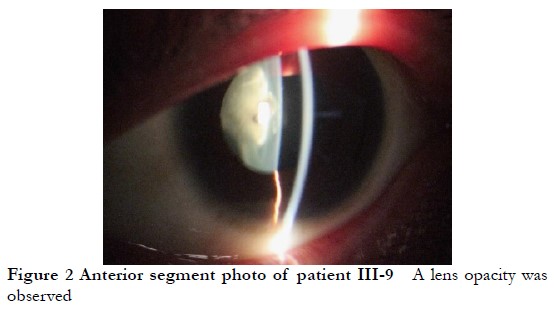
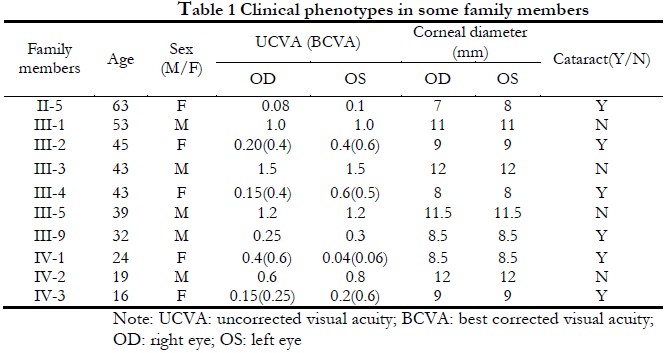
The lineage was consistent with autosomal dominant inheritance (Figure 1). The proband, a female 16-years-old, had poor vision since childhood, and was diagnosed with binocular congenital cataract at Xiamen Eye Center Affiliated to Xiamen University. In 2005, she was treated with phacoemulsification extraction combined with intraocular lens implantation at the Xiamen Eye Center Affiliated to Xiamen University. and in 2017, she underwent episcleral compression of the right eye at the Xiamen Eye Center Affiliated to Xiamen University for retinal detachment. The cornea diameter of both eyes was 9 mm, the axial length of the eye was 22.22 mm in the right eye, 20.73 mm in the left eye, and the diagnosis was CCMC. Among the 39 members of the CCMC family, 11 CCMC patients had poor vision since childhood, microcorneas, and lens opacities (Figure 2) with corneal diameters of 7–9 mm. Other eye examinations and systemic examinations were generally normal. The clinical phenotypes of some members of this family are shown in Table 1.

2.2 Mutation gene detection and analysis
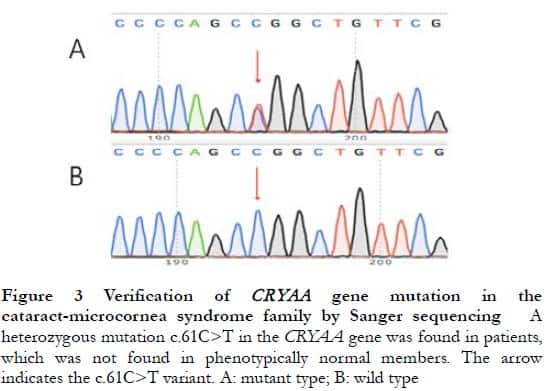
The proband CRYAA gene hybrid mutation, c.61C>T, was found by targeted high-throughput sequencing, which was verified by Sanger sequencing to be present in all patients and not found in normal phenotypic family members, consistent with the co-separation of gene mutations and clinical phenotypes (Figure 3). The dbSNP database search showed that the allele frequency of this variant site in the normal population was extremely low (A < 0.000 01), and InterPro analysis showed that the mutation site was located in the conserved region of the CRYAA gene, causing the 21st amino acid to mutate from arginine (Arg) to tryptophan (Trp). ProtParam analysis showed a decrease in the total number of cationic groups (Arg+Lys) of the mutant protein, and a decrease in hydrophilicity (grand average of hydropathicity: −0.469) and stability with an instability index (II) of 51.54. The PolyPhen-2 online software predicted that the R21W mutation in the CRYAA gene was “likely to impair” protein function with a score of 1.000. The results of the CRYAA gene hybrid mutation c.61C>T homology analysis showed that this locus was highly conserved in monkeys, dogs, cattle, mice, and other species.
3 Discussion
CCMC presents with a corneal diameter less than 11 mm and hereditary cataracts, and other rare ocular manifestations include myopia, abnormal irides, and Peters’ anomaly [15-16]. There is clinical heterogeneity in the cataract phenotype in CCMC in terms of age of onset and progression of the disease. Cataracts in CCMC patients can be divided into star, cortical, karyotype, and mixed type according to the morphology of the lens opacity [4]. In this study, one CCMC family was found in the Youxi area, Fujian, in which the patients had poor vision since childhood, the corneal diameter of all patients was less than 9 mm, there were no other systemic disease, and the cataract type was mixed in unoperated patients. The cornea diameters of the non-affected members were normal.
More than 100 genes have been reported to be associated with congenital cataracts [16], of which about one-half are lens protein genes and one-quarter are membrane protein genes [17]. Crystallin constitutes the major protein of the vertebrate eye lens, with 90% being water-soluble proteins, which play a key role in maintaining lens transparency and refraction [18]. Mutations in crystallin genes may affect protein stability, solubility, and oligomerization properties, and interfere with the orderly arrangement of lens proteins, which leads to cataract formation [19]. According to differences of solubilities, water-soluble lens proteins can be divided into three subcategories: α, β, and γ. The α-crystallin belongs to the family of small molecule heat shock proteins with chaperone function, which maintain the solubility of denatured lens proteins by mimicking chaperones, maintaining lens transparency, and playing an important role in preventing apoptosis and preserving cytoskeletal integrity [20].
This study confirmed that the cause in this CCMC family was heterozygous mutation of c.61C>T of the CRYAA gene, and its expression product, in αA crystallin, the 21st amino acid arginine in the nitrogen terminal region of the lens protein being replaced by tryptophan. Previous studies have also reported that the CRYAA mutation was associated with CCMC [4, 21-22], in which congenital cataracts caused by multiple mutation sites on the CRYAA gene reported by Devi et al. [22] were combined with a microcornea phenotype. These mutations are mainly located in the small heat shock protein conserved area of the N-terminal and a conserved α-crystallin domain region. The CRYAA gene mutation in this study was different from previous studies, and was the first time it has been reported in China. The congenital cataract mutation sites caused by the CRYAA gene are mainly located in the N-terminal and a conserved α-crystallin domain region, which is speculated to be related to the stronger conservatism of the regional sequence and the greater effect on the α-crystallin mimicking chaperone activity after mutation [23], which is consistent with the mutation found in this study. Bioinformatics analyses of mutation products at different sites showed that the total number of cationic groups of protein products was decreased due to the decrease of the total number of cationic groups of protein products and the decrease of hydrophilicity and stability, resulting in the occurrence of cataracts. The results of PolyPhen-2 prediction software suggested that the mutation was a harmful mutation, and homology analysis showed that the mutation site was highly conserved in multiple species.
Genes associated with CCMC are now known to involve CRYAA, CRYBB1, CRYBB2, GRYGC, GRYGD, GJA8, MIP, and MAF [24]. In addition to p.R21W mutations, which cause microcornea, four other site mutations also cause CCMC (Table 2), and in severe cases, microphthalmia [24]. These mutations are all associated with CCMC, but the phenotype that causes cataracts is not identical.

At present, the mechanisms of congenital cataracts combined with microcornea are not clear, and the related hypotheses can be divided into two categories: (1) mutations of transcription factors or growth factors, which play an important role in maintaining and promoting the structural development of the lens and anterior segment of the eye, and the mutated genes can be manifested as pleiotropy [25]; and (2) in addition to inducing and regulating the lens in the embryonic stage, the mutant gene also has a multi-level induction effect on cell differentiation of other anterior eye tissues. Lens dysplasia can indirectly lead to abnormalities in the anterior segment of the eye, including the microcornea. For the existence of two different phenotypes of microcornea and microphthalmia in some gene mutations, it is speculated that the two may belong to the same category of clinical phenotypes, but their severity is different, which may be related to the disorder of eye development induction and regulation caused by lens abnormalities. This is also consistent with the presence of two clinical phenotypes in this family, involving microcornea and microphthalmia. Some researchers have also speculated that it may be caused by corneal development arrest after the fifth month of pregnancy, but at present, the mechanisms of the CRYAA gene mutation and anterior segment developmental abnormalities have not been reported. Further study of these phenotypes, understanding the functions of different genes, and analyzing their role in lens development and their relationship with the anterior segment structure of the eye are expected to explain the role of mutation in the pathogenesis of CCMC.
In this study, the pathogenic mutation of the CCMC family involved the heterozygous mutation, c.61C>T (p.R21W), of the CRYAA gene, which is the first report in China. The microcornea may be associated with disorders of induction regulation due to abnormal lens development. This study has aided our understanding of the role of the CRYAA gene in cataracts and eye development. Further animal experiments of this mutation should help to clarify its pathogenesis.
Conflict of interests None declared.
Author contribution Zhang Daren, Lu Lan: Study conception, manuscript writing and revision; Zeng Jie: Study implementation and data collection; Li Danli, Wang Yun, Wang Xizhen, Fan Ning: Data interpretation, manuscript review; Huang Li: Manuscript writing and revision; Liu Xuyang: Study conception and paper review
References
[1] Wilson ME, Pandey SK, Thakur J. Paediatric cataract blindness in the developing world: surgical techniques and intraocular lenses in the new millennium[J]. Br J Ophthalmol, 2003, 87(1):14-19. DOI: 10.1136/bjo.87.1.14.
[2] He W, Li S. Congenital cataracts: gene mapping[J]. Hum Genet, 2000, 106(1): 1-13. DOI: 10.1007/s004390051002.
[3] Ionides A, Francis P, Berry V, et al. Clinical and genetic heterogeneity in autosomal dominant cataract[J]. Br J Ophthalmol, 1999, 83(7):802-808. DOI: 10.1136/bjo.83.7.802.
[4] Hansen L, Yao W, Eiberg H, et al. Genetic heterogeneity in microcornea-cataract: five novel mutations in CRYAA, CRYGD, and GJA8[J]. Invest Ophthalmol Vis Sci, 2007, 48(9):3937-3944. DOI: 10.1167/iovs.07-0013.
[5] Hejtmancik JF. Congenital cataracts and their molecular genetics[J]. Semin Cell Dev Biol, 2008, 19(2):134-149. DOI: 10.1016/j.semcdb.2007.10.003.
[6] Litt M, Kramer P, LaMorticella DM, et al. Autosomal dominant congenital cataract associated with a missense mutation in the human alpha crystallin gene CRYAA[J]. Hum Mol Genet, 1998, 7(3):471-474. DOI: 10.1093/hmg/7.3.471.
[7] Richter L, Flodman P, Barria von-Bischhoffshausen F, et al. Clinical variability of autosomal dominant cataract, microcornea and corneal opacity and novel mutation in the alpha A crystallin gene (CRYAA)[J]. Am J Med Genet A, 2008, 146A(7):833-842. DOI: 10.1002/ajmg.a.32236.
[8] Zhang LY, Yam GH, Tam PO, et al. An αA-crystallin gene mutation, Arg12Cys, causing inherited cataract-microcornea exhibits an altered heat-shock response[J]. Mol Vis, 2009, 15:1127-1138.
[9] Zhou G, Zhou N, Hu S, et al. A missense mutation in CRYBA4 associated with congenital cataract and microcornea[J]. Mol Vis, 2010, 16:1019-1024.
[10] Willoughby CE, Shafiq A, Ferrini W, et al. CRYBB1 mutation associated with congenital cataract and microcornea[J]. Mol Vis, 2005, 11:587-593.
[11] Zhang L, Fu S, Ou Y, et al. A novel nonsense mutation in CRYGC is associated with autosomal dominant congenital nuclear cataracts and microcornea[J]. Mol Vis, 2009, 15:276-282.
[12] Wang KJ, Wang BB, Zhang F, et al. Novel beta-crystallin gene mutations in Chinese families with nuclear cataracts[J]. Arch Ophthalmol, 2011, 129(3):337-343. DOI: 10.1001/archophthalmol.2011.11.
[13] Devi RR, Vijayalakshmi P. Novel mutations in GJA8 associated with autosomal dominant congenital cataract and microcornea[J]. Mol Vis, 2006, 12:190-195.
[14] Hansen L, Eiberg H, Rosenberg T. Novel MAF mutation in a family with congenital cataract-microcornea syndrome[J]. Mol Vis, 2007, 13:2019-2022.
[15] Green JS, Johnson GJ. Congenital cataract with microcornea and Peters’ anomaly as expressions of one autosomal dominant gene[J]. Ophthalmic Paediatr Genet, 1986, 7(3):187-194. DOI: 10.3109/13816818609004137.
[16] Seitz B, Naumann GO. Bilateral congenital dentiform cataract and extreme microcornea in eyes with uveal colobomas and persistent hyperplastic primary vitreous[J]. Br J Ophthalmol, 1996, 80(4):378-379. DOI: 10.1136/bjo.80.4.378-a.
[17] Bari KJ, Sharma S, Chary K. Structure of G57W mutant of human γS-crystallin and its involvement in cataract formation[J]. J Struct Biol, 2019, 205(3):72-78. DOI: 10.1016/j.jsb.2019.02.003.
[18] Hejtmancik JF, Riazuddin SA, McGreal R, et al. Lens biology and biochemistry[J]. Prog Mol Biol Transl Sci, 2015, 134:169-201. DOI: 10.1016/ bs.pmbts.2015.04.007.
[19] Wistow G. The human crystallin gene families[J/OL]. Hum genomics, 2012, 6:26[2022-03-10]. https://pubmed.ncbi.nlm.nih.gov/23199295/. DOI: 10.1186/ 1479-7364-6-26.
[20] Tikhomirova TS, Selivanova OM, Galzitskaya OV. α-Crystallins are small heat shock proteins: functional and structural properties[J]. Biochemistry (Mosc), 2017, 82(2):106-121. DOI: 10.1134/S0006297917020031.
[21] Hansen L, Mikkelsen A, Nürnberg P, et al. Comprehensive mutational screening in a cohort of Danish families with hereditary congenital cataract[J]. Invest Ophthalmol Vis Sci, 2009, 50(7):3291-3303. DOI: 10.1167/iovs.08-3149.
[22] Devi RR, Yao W, Vijayalakshmi P, et al. Crystallin gene mutations in Indian families with inherited pediatric cataract[J]. Mol Vis, 2008, 14:1157-1170.
[23] Phadte AS, Santhoshkumar P, Sharma KK. Characterization of an N-terminal mutant of αA-crystallin αA-R21Q associated with congenital cataract[J]. Exp Eye Res, 2018, 174:185-195. DOI: 10.1016/j.exer.2018.05.016.
[24] Deng H, Yuan L. Molecular genetics of congenital nuclear cataract[J]. Eur J Med Genet, 2014, 57(2-3):113-122. DOI: 10.1016/j.ejmg.2013.12.006.
[25] Beebe DC, Coats JM. The lens organizes the anterior segment: specification of neural crest cell differentiation in the avian eye[J]. Dev Biol, 2000,220(2):424- 431. DOI: 10.1006/dbio.2000.9638.