•Clinical Research•
Clinical and genetic characteristics of a Han Chinese family with autosomal recessive enhanced S-cone syndrome
Jiang Yongqiang, Chen Kang, Li Jie, Guo Haoyi
Department of Ophthalmology, Henan Provincial People’s Hospital, Henan Eye Hospital, Henan Eye Institute, Zhengzhou 450003, China
Corresponding author: Guo Haoyi, Email: haoyiguo2000@aliyun.com
Abstract [View PDF in English] [View PDF in Chinese] [Read Full Text]
Objective To investigate the clinical symptoms and genetics of a Han Chinese family with enhanced S-cone syndrome (ESCS).
Methods From June to September 2021, we adopted a pedigree investigation approach to examine a Han Chinese family with suspected ESCS. Eight family members from three generations were recruited to Henan Eye Hospital, of which one was suspected with ESCS, and two members had passed away. Eye symptoms of proband were evaluated, including visual acuity, strabismus degree, anterior segment and fundus by using autofluorescence imaging, fluorescein fundus angiography (FFA), full-field electroretinogram (ERG), multifocal ERG, and optical coherence tomography. Peripheral blood samples were taken from the proband and 5 family members to extract DNA, and pathogenic genes and variations were screened using whole exome sequencing (WES). Variations and co-segregation were verified by Sanger sequencing. Variation deleteriousness was analyzed by SIFT, Polyphen2, and MutationTaster methods. Variation pathogenicity was evaluated in accordance with standards and guidelines issued by the American College of Medical Genetics and Genomics (ACMG). Conserved amino acid sequence analyses were performed using SIFT. This study adhered to Declaration of Helsinki principles and the study protocol was approved by an Ethics Committee of Henan Eye Hospital (No. HNEECKY-2017[6]). Written informed consent was obtained from all participants.
Results This pedigree was consistent with an autosomal recessive inheritance mode. The proband displayed some clinical symptoms such as night blindness, hyperopia, accommodative esotropia, peripheral retinal pigmentation, retinoschisis, and photopic ERG responses dominated by large-amplitude waves mediated by S-cone cells. Variations comprising a compound heterozygous variation, c.671C>T: p.S224L on exon 5 and c.955G>A: p.E319K on exon 6 of nuclear receptor subfamily 2, group E, member 3 (NR2E3) were identified by WES. Variations were consistent with co-segregation. Both loci were missense mutations; the mutation frequency of both was 0 in East Asian populations as indicated by the gnomAD database. Variations were predicted as deleterious by SIFT, Polyphen2, and MutationTaster programs. The c.671C>T variation was assigned ‘unknown significance’ in the Clin Var database, and the c.955G>A variation was an unreported new locus. According to ACMG standards and guidelines, both variations were assigned uncertain clinical significance and were highly conserved across multiple species.
Conclusions This proband exhibited the clinical characteristics of ESCS and met diagnostic genetic criteria. Two novel variations c.671C>T: p.S224L and 955G>A: p.E319K in NR2E3 were identified.
[Keywords] Enhanced S-cone syndrome; Pedigree; Whole exome sequencing; Nuclear receptor subfamily 2 group E member 3
Fund program: Henan Medical Science and Technology Project (LHGJ20200069)
DOI: 10.3760/cma.j.cn115989-20210902-00490
Enhanced S-cone syndrome (ESCS) is a rare autosomal recessive retinal disease usually caused by mutation in nuclear receptor subfamily 2, group E, member 3 (NR2E3), which is expressed in photoreceptor cells 1. The major clinical manifestations of ESCS are night blindness, hyperpresbyopia, accommodative esotropia, macular retinoschisis, photopic electroretinogram (ERG) responses, and S-cone-mediated large-amplitude waves. In 1990, Marmor et al. 2 first reported eight ESCS cases. In China, no such case reports exist. In this study, we conducted genetic testing using whole exome sequencing (WES) on a Han Chinese family with suspected ESCS. Pathogenic loci were identified and patient clinical symptoms analyzed, thereby providing a reference point for the clinical diagnosis of ESCS.
1 Materials and methods
1.1 General data
We conducted a pedigree investigation in a Han Chinese family with suspected ESCS. Eight members from three generations were enrolled at the Henan Provincial Eye Hospital from June to September 2021, of which two with normal phenotype had passed away. The clinical data of six families were collected. Five family members were phenotypically normal, and one was suspected with ESCS. Another 60 healthy subjects who received physical examinations at the hospital during the same period, and had no eye and systemic diseases, were enrolled as normal controls. The study was conducted following the principles of the Declaration of Helsinki, and the study protocol was reviewed and approved by an Ethics Committee of Henan Provincial Eye Hospital [Approval No: HNEECKY-2017(6)]. All participants understood the study objectives and voluntarily signed informed consent forms.
1.2 Methods
1.2.1 Clinical examination of the family
We gathered comprehensive information on family, fertility, and systemic disease history, and also marital status. Eye examinations were performed on the proband and phenotypically normal family members. Uncorrected visual acuity and best corrected visual acuity were measured using a logarithmic visual acuity chart. The degree of binocular strabismus was determined using a corneal reflection test, and extraocular muscle functions were examined. Anterior segment and fundus examinations were performed using slit lamp microscopy and binocular indirect ophthalmoscopy, respectively. Retinal autofluorescence was examined using a panoramic fundus camera (OPTOS, UK), visual field was examined using Octopus perimetry (Haag-Streit, Switzerland), and macular structures were examined using Ophthalmic Optical Coherence Tomography v1.33.1 (SVision Imaging, Henan, China). A laser ophthalmic fundus angiography instrument (Heidelberg, Germany) was used for fluorescein fundus angiography (FFA), and a RETI-port visual electrophysiology system was u(Roland, Germany)sed to record full-field ERG. In accordance with International Society for Clinical Electrophysiology of Vision standards (2015) for recording ERG, four scotopic adaptation tests were successively conducted: (1) scotopic 0.01 ERG, light stimulation intensity = 0.01 (cd·s)/m2; (2) scotopic 3.0 ERG, light stimulation intensity = 3.0 (cd·s)/m2; (3) scotopic 3.0 oscillatory potential, light stimulation intensity = 3.0 (cd·s)/m2; and (4) scotopic 10.0 ERG, light stimulation intensity = 10.0 (cd·s)/m2. Following 10 min photopic adaptation, two photopic adaptation tests were performed: (1) photopic 3.0 ERG, light stimulation intensity = 3.0 (cd·s)/m2 and (2) photopic 3.0 flicker ERG, light stimulation intensity = 3.0 (cd·s)/m2. All tests were performed by the same doctor. Both a and b waveforms of mixed light responses, a and b waveforms of photopic ERG responses, and 30 Hz flicker ERG response waveforms were analyzed. Moreover, multifocal ERG (mfERG) was recorded using the RETI-port visual electrophysiology system (Roland, Germany). The mean amplitude density of N1 and P1 waves in the corresponding retinal area of each ring was analyzed using 61 hexagonal stimuli at 47 s per cycle.
1.2.2 WES of pathogenic genes
WES was performed on the proband and her brother, father, maternal grandfather, and uncle, and retinal diseases- and eyeball development-related genes were analyzed.
1.2.2.1 Genomic DNA extraction
From participants, 5 mL peripheral blood was taken and genomic DNA extracted using a magnetic bead blood genomic DNA extraction kit (Tiangen Biotech, Beijing). DNA concentrations and quantities were determined (concentrations were ≥ 50 ng/µL and purity at A260/280: 1.8–2.0 were ≥ 6 µg) using a Qubit 3.0 fluorometer (Qubit double-stranded DNA assay kit, Invitrogen, USA). DNA purity and integrity were also visually assessed using agarose gel electrophoresis.
1.2.2.2 Genome library construction
Eligible genomic DNA fragments were generated using a Covaris M220 Focused-ultrasonicator, and liquid phase capture was performed on whole exome regions using the xGen® Exome Research Panel v2.0 kit (Integrated DNA Technologies, USA), followed by library construction.
1.2.2.3 WES and Sanger sequencing validation
The DNA library was subjected to paired-end sequencing (2×150 bp) of whole exome sequences using the Illumina Hiseq X sequencing platform (Illumina, USA). Original sequencing data were acquired, followed by quality control. Eligible data were compared using the UCSC human genome reference sequence GRCh38 in the Burrows-Wheeler Aligner program (version 0.7.16a, http://bio-bwa. sourceforge.net/). The resultant bam file was assessed for variations, including single nucleotide polymorphisms, insertions, and deletions using the Genome Analysis Tool Kit (version 4.0.8.1). Based on sequencing depth and variation quality, variations were filtered (Quality/Depth ratio > 2 and variation locus depth of at least 20×). Variations in loci were annotated using ANNOVAR (http://annovar.openbioinformatics.org/) to generate candidate pathogenic mutated loci. Then, Sanger sequencing was conducted on all these loci to rule out false positives, and used to determine if loci co-segregated with clinical symptoms. Finally, pathogenic mutated loci were determined.
1.2.3 Pathogenic and conserved loci
Variation deleteriousness was analyzed using SIFT, Polyphen2, and MutationTaster programs, and loci pathogenicity analyzed in accordance with standards and guidelines of the American College of Medical Genetics and Genomics (ACMG) for the Classification of Genetic Variations. Conserved amino acid sequences were analyzed using the SIFT program.
2 Results
2.1 Clinical characteristics of the ESCS pedigree
Autosomal recessive inheritance was verified in this pedigree (Figure 1). Proband III-1, who was 21 years old, had complained of night blindness since childhood, esotropia in near vision, and asthenopia. Optometry results showed the following; +4.50 DS-0.75 DC×5° = 0.7 for the right eye and +5.00 DS-1.25 DC×175° = 0.3 for the left eye. Binocular strabismus ranged from 0° to +10°. The alternate cover test showed internal eyeball and positive movement (Figure 2). Binocular movement examinations showed mild hyperfunction of the right inferior oblique muscle and mild decline in the superior oblique muscle. Horizontal binocular latent nystagmus was also confirmed. No obvious abnormalities of anterior segment were found in both eyes. The bilateral optic discs were normal with clear boundary and color. Pigment-like deposition was observed in the peripheral retina, and light yellow lesions were observed at the vascular arch above the macular area, and retinal pigment epithelium defects had occurred in both eyes (Figure 3). Autofluorescence of the macular area in the vascular arch was enhanced in both eyes (Figure 4). Small hyporeflective cavities between the retinal layers of the outer plexiform layer in the macular area were observed on the image of OCT , showing a worm bite-like appearance (Figure 5). Retinal vessels and the optic disc had normal morphology, and no fluorescein leakage in the macular area was observed by FFA (Figure 6). Annular scotomas and paracentral scotomas were identified in binocular visual field tests (Figure 7). Also, rare characteristic changes had occurred by ERG; ERG could not be recorded upon low-intensity light (0.01) stimulation under scotopic adaptation, while large and slow response waves were recorded upon high-intensity light (3.0) stimulation. The waveform showed no change with increased background light intensity (photopic adaptation), which was distinct to normal ERG responses to long-wavelength light. Moreover, scotopic adaptation was seriously damaged, and retinal rod response was unrecordable. 30 Hz flicker ERG presented very small and delayed waveforms (Figure 8). As indicated by mfERG, the amplitude density of N1 and P1 waves in Ring 1 had decreased slightly, the waveform was normal, and latency was delayed with increased eccentricity. As shown in two-dimension mfERG, the central peak of the left eye shifted to the temporal side, exhibiting paracentral fixation (Figure 9). The proband’s mother (II-2) had died of cerebral hemorrhage in 2018, and her ocular condition was normal before death. The proband’s brothers (III-2 and III-3, 25 and 28 years old, respectively), father (II-3, 52 years old), maternal grandfather (I-1, 78 years old), and uncle (II-1, 56 years old) had a normal bilateral gaze position, and no obvious abnormalities as indicated by fundus examinations and full-field ERG. Thus, pedigree co-segregation was confirmed in this family.
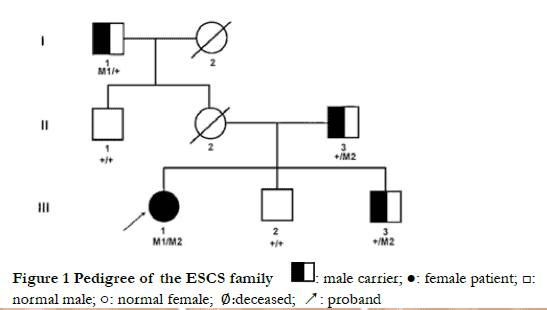

Figure 2 The cardinal position of gaze in proband In nine cardinal positions used to diagnose accommodative esotropia, the reflective spot on cornea appeared in the temporal side of the left pupil. No muscle paralysis signs were observed in other positions. (A) gaze toward upper right (B) upward gaze (C) gaze toward upper left (D) gaze toward right (E) primary position (F) gaze toward left (G) gaze toward down right (H) downward gaze (I) gaze toward down left

Figure 3 Fundus images of proband The optic discs were normal in both eyes with clear boundary and color. Peripheral retinal pigmentation, and yellowish lesions around the vessel arches were observed (A) right eye (B) left eye

Figure 4 Fundus autofluorescence images of proband Autofluorescence was enhanced in the macular area within the vascular arch in both eyes. (A) right eye and (B) left eye

Figure 5 OCT images of proband Dense irregular hyporeflective cavities were observed between retinal layers near the macular outer plexiform layer (A) right eye (B) left eye
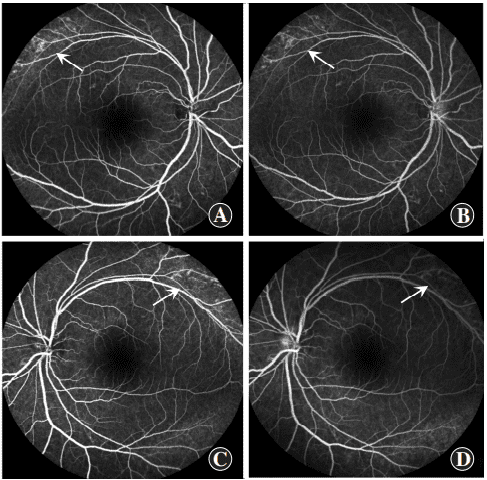
Figure 6 Fluorescein fundus angiography (FFA) images of proband Patchy weak fluorescence (arrows) was observed in the superior temporal beyond the vascular arch in both eyes but not consequentially enhance in the late stage of FFA, and no fluorescein leakage was observed in the macular zone (A and B): right eye (C and D): left eye
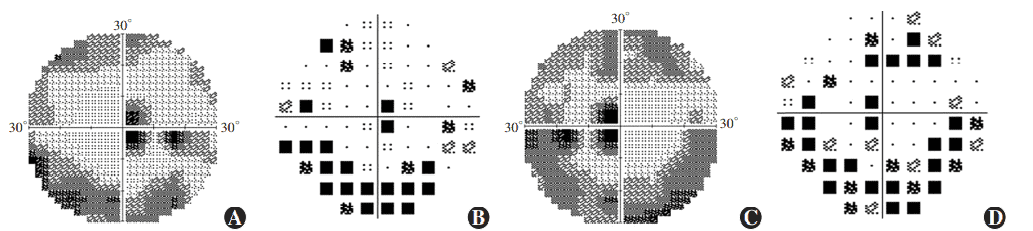
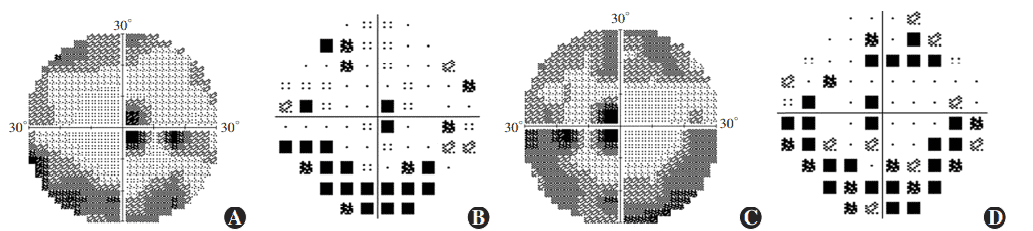
Figure 7 Perimetry results of proband Ring scotomas and paracentral scotomas were found in both eyes (A) Grayscale map of the right eye (B) Pattern deviation map of the right eye (C) Grayscale map of the left eye (D) Pattern deviation map of the left eye

Figure 8 Electroretinogram (ERG) findings of proband (A1, B1): Curve 1 shows maximal responses were unrecordable in the right eye (A1) and left eye (B1) in scotopic 0.01 ERG. Curve 3 displayed moderate to severe reductions of a-wave, moderate decrease of b-wave, and delayed latency of b-wave in scotopic 3.0 ERG. Curve 5 showed a moderate reduction of a- and b-waves and delayed latency in scotopic 10.0 ERG (A2, B2): Mild reduction of a- and b-waves and increased latency were seen in the maximal response in the right eye (A2) and left eye (B2) in photopic 3.0 ERG (A3, B3):Moderately reduced N1-P1 amplitude in the right eye (A3) and left eye (B3) in photopic 3.0 flicker ERG (30 Hz) (C1, C2, C3): Maximal responses, scotopic 3.0 ERG and photopic 3.0 flicker ERG in a normal control
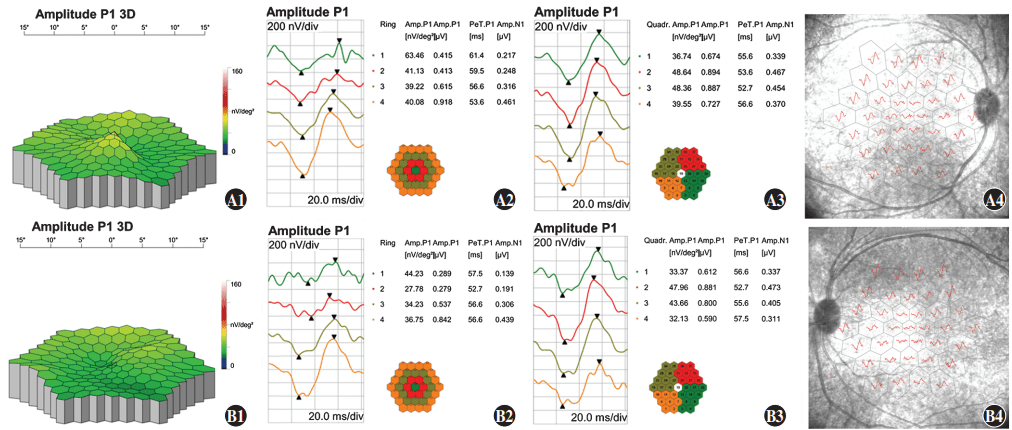
Figure 9 Multifocal electroretinogram (mfERG) findings of proband A1: A three-dimension (3D) display of the P1 wave showing central fixation in the right eye B1: A 3D display of the P1 wave showing paracentral fixation with temporal deviation in the left eye A2, A3, A4, B2, B3, B4: In ring 1, the amplitude density of N1 and P1 waves decreased with a relatively normal waveform, and latency was delayed as centrifugation degree increased A: right eye B: left eye
2.2 Genetic testing of the ESCS pedigree
The proband had a compound heterozygous variation in NR2E3 gene, and two loci variations were identified as missense mutations and comprised c.671C>T on exon 5 and c.955G>A on exon 6. The former changed serine to leucine at position 224 of the NR2E3 protein, while the latter altered glutamic acid to lysine at position 319 (Figure 10). Using gnomAD, the c.671C>T variation frequency in East Asian populations was 0. It was recorded as ‘undetermined significance’ in ClinVar and was not reported in the literature. The c.955G>A variation was not included in East Asian populations in gnomAD and ClinVar databases, and was similarly unreported in the literature. Both variations were deleterious as predicted by SIFT, Polyphen2, and MutationTaster programs, and were of undetermined clinical significance based on ACMG standards and guidelines. From pedigree analyses, the c.955G>A variation was inherited from the proband’s father, but the source of the c.671C>T variation could not be determined as the proband’s mother was deceased. Comparative analysis of NR2E3 amino acid sequences between Homo sapiens, Bos taurus, Pan troglodytes, Mus musculus, Danio rerio, and Gallus gallus showed that amino acids 224 and 319 were highly conserved across species (Figure 11), and that pathogenicity after mutation was high. When combined with clinical symptoms, ESCS caused by mutated NR2E3 was diagnosed.
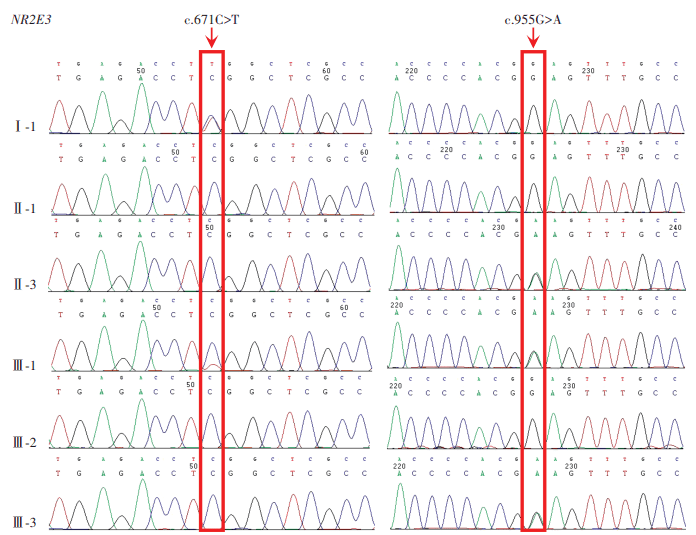 Figure 10 Sanger sequencing map of the ESCS family A compound he terozygous variation c.671C>T/c.955G>A was identified in NR2E3 in the proband
Figure 10 Sanger sequencing map of the ESCS family A compound he terozygous variation c.671C>T/c.955G>A was identified in NR2E3 in the proband
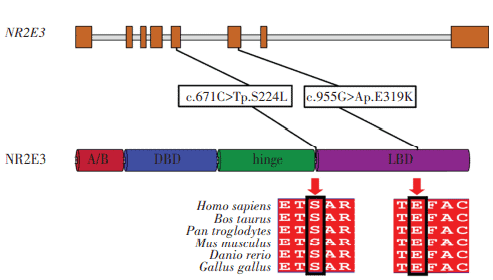
Figure 11 NR2E3 structure and conservation across species analyses NR2E3 amino acid positions at 224 and 319 were highly conserved across Homo sapiens, Bos taurus, Pan troglodytes, mus musculus, Danio rerio, and Gallus gallus species
3 Discussion
At the age of 2 years, the proband was diagnosed with night blindness. After admission to our hospital, ERG and OCT showed that retinal rod and cone functions were damaged, multiple small retinoschisis chambers were present between macular retinal layers, accommodative esotropia was observed in both eyes, and scotopic adaptation was severely impaired, consistent with ESCS. Gene sequencing indicated the proband had two mutated loci at c.671C>T (p.S224L) and c.955G>A (p.E319K) in NR2E3, which were not previously reported in the literature. When combined with clinical symptoms, ESCS was confirmed in the proband and was caused by a compound heterozygous variation.
In total, 78 ESCS-related eye disease studies were retrieved from PubMed. In studies reporting ESCS caused by NR2E3 mutations, 84 mutated loci were listed, including 58 missense, 15 splicing, six frameshift, and four deletion mutations, and also one nonsense mutation. Amongst mutations, three were located in the N-terminal A/B domain, 24 in the DNA binding domain, five in the hinge region, and 37 in the ligand binding domain. We observed that c.119-2A>C and c.932G>A (p.R311Q) were two commonly mutated loci 3-29.
ESCS is an autosomal recessive retinal dystrophy caused by mutations in the nuclear receptor gene NR2E3 on chromosome 15q23. In childhood, patients usually present with normal fundus or a few discrete white spots in the retinal pigment epithelium, retinal mass pigmentation is usually seen at 9–11 years old and also macular cystoid changes not accompanied by fluorescein leakage or cleavage by FFA 30-31. Three cone photoreceptor types exist in the human retina: short-wave-sensitive (S, blue), medium-wave-sensitive (M, green), and long-wave-sensitive (L, red) cones. In all types, inherited retinal dystrophy generates progressive rod and cone cell degeneration. However, ESCS has enhanced short-wave cone function, severe medium- and long-wave cone function impairment, and characteristic rod function changes. Based on such pathophysiological changes, rare characteristic changes can be identified by ERG in such patients, i.e., ERG cannot be recorded upon low-intensity light (0.01) stimulation under scotopic adaptation, while large and slow response waves are recorded upon high-intensity light (3.0) stimulation, and waveforms show no changes with increased background light intensity (photopic adaptation), distinct to normal ERG responses to long-wavelength light.
NR2E3 is specifically expressed in photoreceptor cells and was identified due to its high conservation with the protein sequence and structure of NR2E1. NR2E1 is implicated in cell fate determination mechanisms during Drosophila forebrain and eye development, and encodes an orphan receptor of the steroid/thyroid hormone receptor superfamily of ligand-activated transcription factors 33-34. Mutations in human NR2E3 may cause several retinal diseases such as autosomal dominant and recessive retinitis pigmentosa, Goldmann-Favre syndrome, clumped pigmentary retinal degeneration, and ESCS 17, 35-40. ESCS shares some symptom similarities with Goldmann-Favre syndrome, which is considered a serious version of ESCS 40. NR2E3 encodes a 410 amino acid polypeptide (relative molecular mass = 45.5 kDa) and contains an amino-terminal A/B domain, DNA binding domain, hinge region, and carboxyl-terminal ligand binding domain. The DNA binding domain binds to target gene promoters, while the ligand binding domain binds to as yet unknown ligands, and the hinge region is the nuclear localization signal 9,41-42. In NR2E3-deficient rd7 M. musculus, rod cell development, differentiation, and maturation are normal 1,43-46. NR2E3 is conserved in rod cell development/regulation in H. sapiens and D. rerio. Rod precursor cells cannot differentiate into mature rod cells in D. rerio expressing a NR2E3 mutation, while rod cell functions in ESCS patients are severely damaged at early disease stages 47, resulting in early night blindness.
In early stage of ESCS, congenital static night blindness and non-pigmented retinitis pigmentosa has some similar manifestations, such as poor night vision, mild to moderate visual loss, and no serious abnormalities in peripheral and normal visual fields, therefore, these diseases are difficult to distinguish only by chief complaint and fundus phenotype, but all have different ERG characteristics. During clinical diagnoses, it is vital to understand the ERG characteristics of retinopathies with similar early symptoms. ERG examinations should be considered when the disease type cannot be clearly identified by the patient, and a definitive diagnosis should be made in combination with genetic testing results, thereby facilitating targeted treatment and improving the patient’s quality of life. The phenotypic heterogeneity of ESCS is high, causal relationships between retinal cell degeneration and NR2E3 mutations are unclear, and phenotypes following NR2E3 mutation vary across different species. Consequently, it is difficult to comprehensively determine NR2E3 functions, therefore no effective clinical treatments for the condition have been identified. In our pedigree, the proband is 21 years old, and a long-term follow-up observation to monitor fundus disease development is available to contribute to ESCS genetic analyses.
In this study, two unreported mutated loci c.671C>T:p.S224L and c.955G>A:p.E319K were identified in NR2E3. Our study has enriched the gene mutation spectrum of autosomal recessive ESCS, generated comprehensive insights on ESCS pathogenesis, and provided a reference point for clinical genetic counseling and fertility guidance.
Conflict of interest The authors declare none.
Author’s contribution statement Jiang Yongqiang: topic selection, test design, study implementation, and paper writing; Guo Haoyi: test design, review, and revision of the intellectual content of the article; Li Jie and Chen Kang: data collection, data sorting, and analysis
References
[1] Haider NB, Demarco P, Nystuen AM, et al. The transcription factor Nr2e3 functions in retinal progenitors to suppress cone cell generation[J]. Vis Neurosci, 2006, 23(6):917-929. DOI: 10.1017/S095252380623027X.
[2] Marmor MF, Jacobson SG, Foerster MH, et al. Diagnostic clinical findings of a new syndrome with night blindness, maculopathy, and enhanced S cone sensitivity[J]. Am J Ophthalmol, 1990, 110(2):124-134. DOI: 10.1016/s0002-9394(14)76980-6.
[3] Jurklies B, Weismann M, Kellner U, et al. Clinical findings in autosomal recessive syndrome of blue cone hypersensitivity[J]. Ophthalmologe, 2001, 98(3):285-293. DOI: 10.1007/s003470170164.
[4] Bernal S, Solans T, Gamundi MJ, et al. Analysis of the involvement of the NR2E3 gene in autosomal recessive retinal dystrophies[J]. Clin Genet, 2008, 73(4):360-366. DOI: 10.1111/j.1399-0004.2008.00963.x.
[5] Kuniyoshi K, Hayashi T, Sakuramoto H, et al. Novel mutations in enhanced S-cone syndrome[J/OL]. Ophthalmology, 2013, 120(2):431-436[2022-02-19]. https://pubmed.ncbi.nlm.nih.gov/23374571/. DOI: 10.1016/j.ophtha.2012.08.032.
[6] Audo I, Michaelides M, Robson AG, et al. Phenotypic variation in enhanced S-cone syndrome[J]. Invest Ophthalmol Vis Sci, 2008, 49(5):2082-2093. DOI: 10.1167/iovs.05-1629.
[7] Coppieters F, Héon E, Cima I, et al. Exploring the role of genes of the retinal transcription network in retinal degeneration[J]. Invest Ophthalmol Vis Sci, 2009, 50(13): 3745-3745.
[8] Coppieters F, Leroy BP, Beysen D, et al. Recurrent mutation in the first zinc finger of the orphan nuclear receptor NR2E3 causes autosomal dominant retinitis pigmentosa[J]. Am J Hum Genet, 2007, 81(1):147-157. DOI: 10.1086/518426.
[9] Park SP, Hong IH, Tsang SH, et al. Disruption of the human cone photoreceptor mosaic from a defect in NR2E3 transcription factor function in young adults[J]. Graefes Arch Clin Exp Ophthalmol, 2013, 251(10):2299-2309. DOI: 10.1007/s00417-013-2296-5.
[10] de Carvalho ER, Robson AG, Arno G, et al. Enhanced S-cone syndrome: spectrum of clinical, imaging, electrophysiologic, and genetic findings in a retrospective case series of 56 patients[J]. Ophthalmol Retina, 2021, 5(2):195-214. DOI: 10.1016/j.oret.2020.07.008.
[11] Haider NB, Jacobson SG, Cideciyan AV, et al. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate[J]. Nat Genet, 2000, 24(2):127-131. DOI: 10.1038/72777.
[12] Rocha-Sousa A, Hayashi T, Gomes NL, et al. A novel mutation (Cys83Tyr) in the second zinc finger of NR2E3 in enhanced S-cone syndrome[J]. Graefe’s Arch Clin Exp Ophthalmol, 2011, 249(2):201-208. DOI: 10.1007/s00417-010-1482-y.
[13] Wright AF, Reddick AC, Schwartz SB, et al. Mutation analysis of NR2E3 and NRL genes in enhanced S cone syndrome[J/OL]. Hum Mutat, 2004, 24(5):439[2022-02-19]. https://pubmed.ncbi.nlm.nih.gov/15459973/. DOI: 10.1002/humu.9285.
[14] Hull S, Arno G, Sergouniotis PI, et al. Clinical and molecular characterization of enhanced S-cone syndrome in children[J]. JAMA Ophthalmol, 2014, 132(11):1341-1349. DOI: 10.1001/jamaophthalmol.2014.2343.
[15] Hayashi T, Gekka T, Goto-Omoto S, et al. Novel NR2E3 mutations (R104Q, R334G) associated with a mild form of enhanced S-cone syndrome demonstrate compound heterozygosity[J/OL]. Ophthalmology, 2005, 112(12):2115[2022-02-20]. https://pubmed.ncbi.nlm.nih.gov/16225923/. DOI: 10.1016/j.ophtha.2005.07.002.
[16] Cehajic-Kapetanovic J, Cottriall CL, Jolly JK, et al. Electrophysiological verification of enhanced S-cone syndrome caused by a novel c.755T>C NR2E3 missense variant[J]. Ophthalmic Genet, 2019, 40(1):29-33. DOI: 10.1080/13816810.2018.1547912.
[17] Sharon D, Sandberg MA, Caruso RC, et al. Shared mutations in NR2E3 in enhanced S-cone syndrome, Goldmann-Favre syndrome, and many cases of clumped pigmentary retinal degeneration[J]. Arch Ophthalmol, 2003, 121(9):1316-1323. DOI: 10.1001/archopht.121.9.1316.
[18] Lam BL, Goldberg JL, Hartley KL, et al. Atypical mild enhanced S-cone syndrome with novel compound heterozygosity of the NR2E3 gene[J]. Am J Ophthalmol, 2007, 144(1):157-159. DOI: 10.1016/j.ajo.2007.03.012.
[19] Termühlen J, Alex AF, Glöckle N, et al. A new mutation in enhanced S-cone syndrome[J/OL]. Acta Ophthalmol, 2018, 96(4):e539-e540[2022-02-20]. https://pubmed.ncbi.nlm.nih.gov/27573156/. DOI: 10.1111/aos.13205.
[20] Nowilaty SR, Alsalamah AK, Magliyah MS, et al. Incidence and natural history of retinochoroidal neovascularization in enhanced S-cone syndrome[J]. Am J Ophthalmol, 2021, 222:174-184. DOI: 10.1016/j.ajo.2020.09.010.
[21] Al-Khuzaei S, Broadgate S, Halford S, et al. Novel pathogenic sequence variants in NR2E3 and clinical findings in three patients[J/OL]. Genes (Basel), 2020, 11(11):1288[2022-02-21]. https://pubmed.ncbi.nlm.nih.gov/33138239/. DOI: 10.3390/genes11111288.
[22] Nakamura Y, Hayashi T, Kozaki K, et al. Enhanced S-cone syndrome in a Japanese family with a nonsense NR2E3 mutation (Q350X)[J]. Acta Ophthalmol Scand, 2004, 82(5):616-622. DOI: 10.1111/j.1600-0420.2004.00328.x.
[23] Pachydaki SI, Klaver CC, Barbazetto IA, et al. Phenotypic features of patients with NR2E3 mutations[J]. Arch Ophthalmol, 2009, 127(1):71-75. DOI: 10.1001/archophthalmol.2008.534.
[24] Minnella AM, Pagliei V, Savastano MC, et al. Swept source optical coherence tomography and optical coherence tomography angiography in pediatric enhanced S-cone syndrome: a case report[J/OL]. J Med Case Rep, 2018, 12(1):287[2022-02-21]. https://pubmed.ncbi.nlm.nih.gov/30285900/. DOI: 10.1186/s13256-018-1819-4.
[25] Sustar M, Perovšek D, Cima I, et al. Electroretinography and optical coherence tomography reveal abnormal post-photoreceptoral activity and altered retinal lamination in patients with enhanced S-cone syndrome[J]. Doc Ophthalmol, 2015, 130(3):165-177. DOI: 10.1007/s10633-015-9487-9.
[26] Ammar MJ, Scavelli KT, Uyhazi KE, et al. Enhanced s-cone syndrome: visual function, cross-sectional imaging, and cellular structure with adaptive optics ophthalmoscopy[J]. Retin Cases Brief Rep, 2021, 15(6):694-701. DOI: 10.1097/ICB.0000000000000891.
[27] Bandah D, Merin S, Ashhab M, et al. The spectrum of retinal diseases caused by NR2E3 mutations in Israeli and Palestinian patients[J]. Arch Ophthalmol, 2009, 127(3):297-302. DOI: 10.1001/archophthalmol.2008.615.
[28] Di Scipio M, Tavares E, Deshmukh S, et al. Phenotype driven analysis of whole genome sequencing identifies deep intronic variants that cause retinal dystrophies by aberrant exonization[J/OL]. Invest Ophthalmol Vis Sci, 2020, 61(10):36[2022-02-23]. https://pubmed.ncbi.nlm.nih.gov/32881472/. DOI: 10.1167/iovs.61.10.36.
[29] Kuniyoshi K, Hayashi T, Sakuramoto H, et al. New truncation mutation of the NR2E3 gene in a Japanese patient with enhanced S-cone syndrome[J]. Jpn J Ophthalmol, 2016, 60(6):476-485. DOI: 10.1007/s10384-016-0470-0.
[30] Udar N, Small K, Chalukya M, et al. Developmental or degenerative–NR2E3 gene mutations in two patients with enhanced S cone syndrome[J]. Mol Vis, 2011, 17:519-525.
[31] Sohn EH, Chen FK, Rubin GS, et al. Macular function assessed by microperimetry in patients with enhanced S-cone syndrome[J]. Ophthalmology, 2010, 117(6):1199-1206.e1. DOI: 10.1016/j.ophtha.2009.10.046.
[32] Zuo CG, Xing YQ, Chen CZ. Enhanced S-cone syndrome[J]. Chin J Ocul Fundus Dis, 2007, 23(3):228-230. DOI: 10.3760/j.issn:1005-1015.2007.03.028.
[33] Kobayashi M, Hara K, Yu RT, et al. Expression and functional analysis of Nr2e3, a photoreceptor-specific nuclear receptor, suggest common mechanisms in retinal development between avians and mammals[J]. Dev Genes Evol, 2008, 218(8):439-444. DOI: 10.1007/s00427-008-0232-1.
[34] Kanda A, Swaroop A. A comprehensive analysis of sequence variants and putative disease-causing mutations in photoreceptor-specific nuclear receptor NR2E3[J]. Mol Vis, 2009, 15: 2174-2184.
[35] Escher P, Tran HV, Vaclavik V, et al. Double concentric autofluorescence ring in NR2E3-p.G56R-linked autosomal dominant retinitis pigmentosa[J]. Invest Ophthalmol Vis Sci, 2012, 53(8):4754-4764. DOI: 10.1167/iovs.11-8693.
[36] Gerber S, Rozet JM, Takezawa SI, et al. The photoreceptor cell-specific nuclear receptor gene (PNR) accounts for retinitis pigmentosa in the Crypto-Jews from Portugal (Marranos), survivors from the Spanish Inquisition[J]. Hum Genet, 2000, 107(3):276-284. DOI: 10.1007/s004390000350.
[37] FAVRE M. Two cases of hyaloid-retinal degeneration[J]. Ophthalmologica, 1958, 135(5-6):604-609. DOI: 10.1159/000303360.
[38] Fishman GA, Jampol LM, Goldberg MF. Diagnostic features of the Favre-Goldmann syndrome[J]. Br J Ophthalmol, 1976, 60(5):345-353. DOI: 10.1136/bjo.60.5.345.
[39] Chavala SH, Sari A, Lewis H, et al. An Arg311Gln NR2E3 mutation in a family with classic Goldmann-Favre syndrome[J]. Br J Ophthalmol, 2005, 89(8):1065-1066. DOI: 10.1136/bjo.2005.068130.
[40] Schorderet DF, Escher P. NR2E3 mutations in enhanced S-cone sensitivity syndrome (ESCS), Goldmann-Favre syndrome (GFS), clumped pigmentary retinal degeneration (CPRD), and retinitis pigmentosa (RP)[J]. Hum Mutat, 2009, 30(11):1475-1485. DOI: 10.1002/humu.21096.
[41] Kobayashi M, Takezawa S, Hara K, et al. Identification of a photoreceptor cell-specific nuclear receptor[J]. Proc Natl Acad Sci U S A, 1999, 96(9):4814-4819. DOI: 10.1073/pnas.96.9.4814.
[42] Mangelsdorf DJ, Thummel C, Beato M, et al. The nuclear receptor superfamily: the second decade[J]. Cell, 1995, 83(6):835-839. DOI: 10.1016/0092-8674(95)90199-x.
[43] Chen J, Rattner A, Nathans J. The rod photoreceptor-specific nuclear receptor Nr2e3 represses transcription of multiple cone-specific genes[J]. J Neurosci, 2005, 25(1):118-129. DOI: 10.1523/JNEUROSCI.3571-04.2005.
[44] Corbo JC, Cepko CL. A hybrid photoreceptor expressing both rod and cone genes in a mouse model of enhanced S-cone syndrome[J/OL]. PLoS Genet, 2005, 1(2):e11[2022-02-24]. https://pubmed.ncbi.nlm.nih.gov/16110338/. DOI: 10.1371/journal.pgen.0010011.
[45] Haider NB, Mollema N, Gaule M, et al. Nr2e3-directed transcriptional regulation of genes involved in photoreceptor development and cell-type specific phototransduction[J]. Exp Eye Res, 2009, 89(3):365-372. DOI: 10.1016/j.exer.2009.04.006.
[46] Ueno S, Kondo M, Miyata K, et al. Physiological function of S-cone system is not enhanced in rd7 mice[J]. Exp Eye Res, 2005, 81(6):751-758. DOI: 10.1016/j.exer.2005.04.013.
[47] Xie S, Han S, Qu Z, et al. Knockout of Nr2e3 prevents rod photoreceptor differentiation and leads to selective L-/M-cone photoreceptor degeneration in zebrafish[J]. Biochim Biophys Acta Mol Basis Dis, 2019, 1865(6):1273-1283. DOI: 10.1016/j.bbadis.2019.01.022.