Genotypes and ocular and systemic clinical phenotypes of the Hermansky-Pudlak syndrome
Yang Shangying1, Cheng Wanyu1, Zhang Yan2, Sheng Xunlun1,3
1Ningxia Eye Hospital, People’s Hospital of Ningxia Hui Autonomous Region, Yinchuan 750001, China; 2Electron Microscope Room, Science and Technology Center, Ningxia Medical University, Yinchuan 750001, China; 3Gansu Aier Optometry Hospital, Lanzhou 730000, China
Corresponding author: Sheng Xunlun, Email: shengxunlun@163.com
[Abstract] [View PDF in English] [View PDF in Chinese] [Read Full Text]
Objective This study aimed to analyze the genotypes and ocular and systemic clinical phenotypes of two families with Hermansky-Pudlak syndrome (HPS).
Methods The method of pedigree investigation was adopted. The clinical data of two probands and their phenotypically normal parents were collected. Relevant ophthalmologic and systemic examinations were carried out. The platelet dense granules in the two probands were observed using an electron microscope. DNA was extracted from the peripheral venous blood collected from the patients. The pathogenic genes were screened by whole-exome sequencing. The potential disease-causing variations were analyzed through bioinformatics analysis. The validation and family cosegregation analysis of the pathogenic variations were performed by Sanger sequencing. The relationship between HPS-related gene variations and clinical characteristics was explored.
Results The two families were consistent with the autosomal recessive inheritance pattern. In family 1 with a family history of consanguineous marriage, the proband had no obvious hypopigmentation on his facial skin, hair, eyebrows, and eyelashes. Horizontal nystagmus, exotropia, mild visual impairment, iris atrophy, positive light transmission, orange fundus, pigment loss, macular hypoplasia, prolonged prothrombin time in laboratory examination, and a significant reduction in the number of platelet dense granules were observed using an electron microscope. The proband in family 2 had pale brown hair and eyebrows, severe visual impairment, normal iris pigment, longer thrombin time in laboratory tests, and other characteristics similar to those of the proband in family 1. A novel homozygous variant c.2887G>T (p.E963X) was detected in the HPS3 gene of the proband in family 1. The parents of the proband from family 1 both carried a heterozygous variant c.2887G>T (p.E963X). Compound heterozygous variants were detected in the HPS5 gene of the proband in family 2, including c.2952-2A>C splicing variation and heterozygous deletion (a 3 144-bp deletion, located in chr11:18302108-18305251, exon 22). The parents of the proband from family 2 carried a heterozygous variation. The three novel variations were labeled as pathogenic according to the American College of Medical Genetics and Genomics Standards and Guidelines.
Conclusions Family 1 had the HPS-3 subtype, and family 2 had the HPS-5 subtype. A certain genotype-phenotype correspondence existed between the two types of HPS.
[Key words] Genetic testing; genotype; Hermansky-Pudlak syndrome; HPS3 gene; HPS5 gene; pedigree; phenotype; whole-exome sequencing
Fund program: National Natural Science Foundation of China (81760180) and the Key R&D project of Ningxia Autonomous Region (2021BEG02045)
DOI: 10.3760/cma.j.cn115989-20210728-00430
The Hermansky-Pudlak syndrome (HPS, MIM 203300) is a kind of albinism syndrome, which is autosomal recessive, with obvious genetic heterogeneity and clinical phenotype heterogeneity. HPS has 11 subtypes, namely HPS-1–HPS-11. HPS-1 and HPS-3 are common subtypes, and HPS-5 is relatively rare 1-3. HPS has a variety of clinical manifestations, mainly characterized by the triad of oculocutaneous albinism (OCA), bleeding tendency, and wax-like lipid accumulation in tissues, with or without fatal complications, such as pulmonary fibrosis, granulomatous colitis, renal failure, and cardiomyopathy. These characteristics are due to the defects in lysosome-related organelles, such as the lack or large reduction of melanosomes in melanocytes and platelet dense particles in platelets. At present, the Human Gene Mutation Database (HGMD) has included more than 200 HPS gene variants. The proteins encoded by different HPS genes belong to different complexes and have different mechanisms of action. However, different subtypes that affect the same organelle biosynthesis complex (BLOC) or adaptor complex-3 (AP-3) often have similar clinical phenotypes, and different subtypes have different severity and different clinical phenotypes, posing great challenges to the differential diagnosis by clinicians. In this study, whole-exome sequencing was used to screen the pathogenic genes of two families with HPS, and the genotype and clinical phenotype characteristics of HPS-related eye diseases were analyzed in combination with previous reports. The purpose was to summarize the relationship between genotype and phenotype of this disease, and to help clinicians improve their understanding of this disease.
1 Data and Methods
1.1 General information
The clinical data of probands and their parents were collected using the method of pedigree investigation, including one Chinese Han and one Hui HPS pedigree who visited the People’s Hospital of Ningxia Hui Autonomous Region from June 2020 to May 2021. This study was conducted in accordance with the Helsinki Declaration. The research scheme was approved by the ethics medical committee of the People’s Hospital of Ningxia Hui Autonomous Region (Approval No.: 2016018). Informed consent was obtained from patients and their families for all genetic testing and diagnosis work, and they signed the consent form voluntarily.
1.2 Methods
1.2.1 Clinical examination of the family members The facial appearance photograph, best-corrected visual acuity (BCVA), slit-lamp microscope examination, color vision, color fundus photograph (TRC-NW300, Topcon, Japan), optical coherence tomography imaging (OCT, HD-OCT4000, Carl Zeiss Meditec, USA), electroretinogram record, visual evoked potential, and fundus fluorescein angiography were examined to collect the relevant clinical phototypes of the probands and their parents. The laboratory coagulation function and pulmonary computed tomography (CT) were performed.
1.2.2 Observation of platelet dense particles of the proband The electron microscopy was completed by experienced experts from the Electron Microscope Room of the Science and Technology Center of Ningxia Medical University. First, 4 mL of peripheral venous blood was collected from the patient in a blood collection vessel containing sodium citrate and glucose solution. It was placed in a 12-cm radius centrifuge and centrifuged at 1,500 rpm for 10 min. Thus, platelets rich in plasma were obtained. The platelet slices were obtained using the embedding method. The morphological changes in platelets and platelet dense granules were observed by transillumination. The gold standard for diagnosing HPS was the absence or large reduction of platelet dense granules observed under an electron microscope 4.
1.2.3 Whole-exon sequencing The whole-exon sequencing of the probands and their parents was performed at the same time (the trio whole-exon group-sequencing mode). An Agilent SureSelect exon capture kit (Santa Clara, California, USA) was used for the whole-genome exon capture, and a high-throughput sequencer (Illumina, California, USA) was used for sequencing. The original sequencing data were processed using Illumina base calling software 1.7 and compared with the National Center for Biotechnology (NCBI) human genome DNA reference sequence (NCBI build 37.1). The SOAP software was used (http://soap.genomics.org.cn) to obtain information related to single-nucleotide variation, and the BWA software (http://bio-bwa.sourceforge.net/) was used to analyze the relevant information of insertion/deletion variation, so as to master all the variations in DNA sequence in the sample. The quality control of the sequencing data was carried out according to the sequencing depth, the ratio of the number of variant reads to the total number of reads at the site, whether it was in the high GC region, or the repeat sequence region, or the poly region. The frequencies of 1KG gnomAD and other databases were screened and more than 20 bioinformatics software were for predicting whether phenotypic genes were cosegregated to identify the relationship between the gene where the mutation was located and the disease. After gradual filtering, the number of variants shared by all patients in the family was screened, and then the variants in the family without diseased relatives were filtered. Finally, the mutation annotation and hazard prediction were conducted to obtain candidate pathogenic gene variants. Sanger validation and co-separation analysis were carried out for suspected pathogenic variants. In addition, polymerase chain reaction (PCR) detection was supplemented for deletion mutation.
1.2.4 Pathogenicity analysis of gene variations Using the HGMD, dbSNP (https://www.ncbi.nlm.nih.gov/snp/), the target mutation site was queried in the database and it was checked whether it was a reported pathogenic mutation and included in HGMD. If it was a new variation that had not been reported, the pathogenicity of the new variation was evaluated according to the Standards and Guidelines for Interpretation of Sequence Variation issued by the American College of Medical Genetics and Genomics (ACMG) in 2015. The online tool LRT (http://www.genetics.wustl.edu/jflab/lrt_query.html) was used and 1000 Genomes (http://browser.1000genomes.org/index.html), Exome Variant Server (EVS, http://evs.gs.washington.edu/EVS/), and Exome Aggregation Consertium (ExAC, http://exac.broadinstitute.org) databases were selected to view the allele frequency of the variation in the normal population.
2 Results
2.1 Clinical phenotype of probands from the two families with HPS
Family 1 The proband, a 21-year-old man, had exotropia, poor vision, and photophobia for more than 10 years. His parents had a family history of consanguineous marriage. His facial skin, hair, eyebrows, and eyelashes had no obvious hypopigmentation. The BCVA of both eyes was 0.3 (right eye: -1.50 DS/-2.00 DC ´ 25°; left eye: -1.75 DS/-2.75 DC ´ 155°), with exotropia -15°, horizontal nystagmus, and normal red and green color vision. He had iris atrophy in both eyes, with positive light transmission in the iris. The fundus of both eyes was orange, with no pigment, and the choroidal large vessels were visible. The OCT indicated that the fovea of macula was poorly developed in both eyes (Figure 1). The laboratory examination only slightly increased the prothrombin time, which was 14.00 s. No obvious abnormality was found in the lung CT examination.
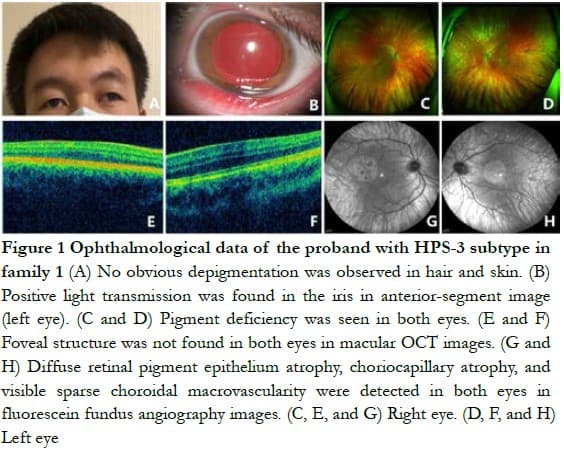
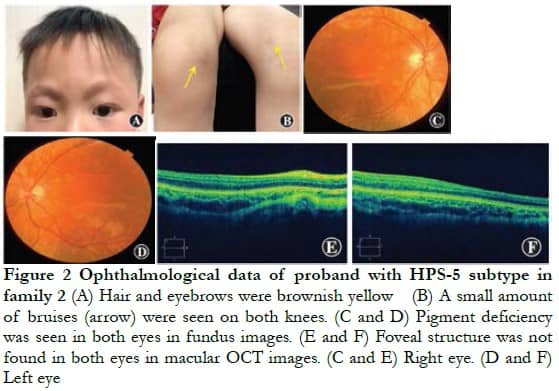
Family 2 The proband, a 4-year-old man, had brownish yellow hair and eyebrows, normal skin color, and a small amount of bruising on both knees. The BCVA of both eyes was 0.04 (right eye: -4.00 DS/-5.00 DC ´ 5°; left eye: -3.00 DS/4.00 DC ´ 85°), color vision could not be matched, the anterior segment of both eyes was normal, and fundus performance was similar to that of the proband of family 1 (Figure 2). The laboratory examination only slightly increased the thrombin time, which was 19.90 s. No obvious abnormality was found in the lung CT examination.
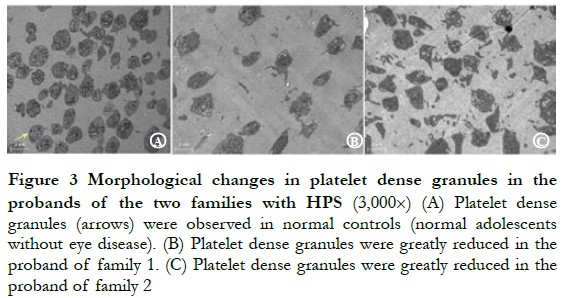
2.2 Morphological changes in platelet dense granules in probands of the two families with HPS
The probands of pedigree 1 and 2 all showed a large reduction in the number of platelet dense granules (Figure 3).
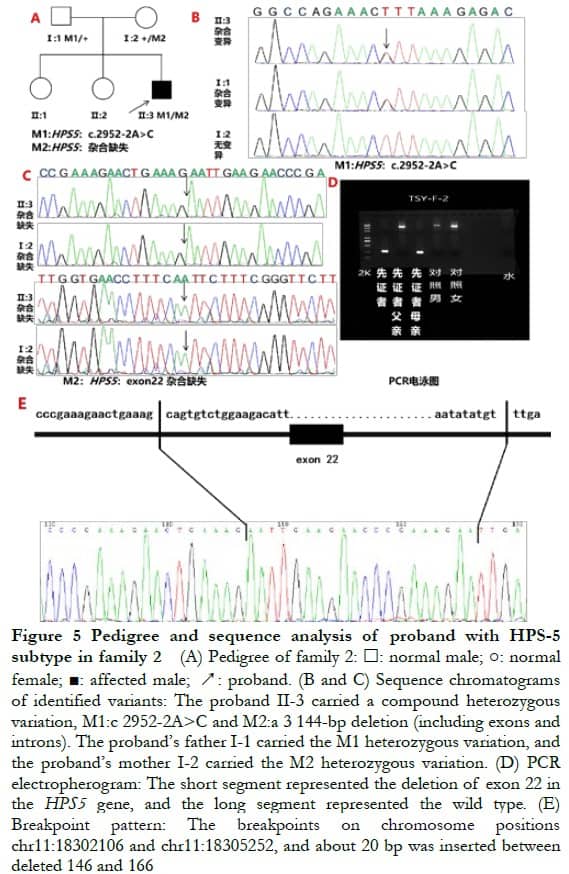
2.3 Full-exon sequencing and bioinformatics analysisA new homozygous nonsense variation of c.2887G>T (p.E963X) was detected in exon 16 of the HPS3 gene of the proband of family 1, and parents with normal phenotype carried c.2887G>T (p.E963X) heterozygous variation, indicating that the genotype and clinical phenotype were separated. This family conformed to autosomal recessive inheritance. Combined with the clinical phenotype of the patient, the preliminary diagnosis was HPS-3 subtype (Figure 4). Compound heterozygous variation was detected in the HPS5 gene of the proband in family 2: c.2952-2A>C splice mutation and loss of heterozygosity (3 144-bp deletion, located at chr11: 18302108–18305251, exon 22). Parents with normal phenotypes carried one heterozygous variant. Sanger sequencing verified that the splice site variation came from the father and the deletion variation came from the mother, which was consistent with the autosomal recessive inheritance mode. Combined with the patient’s clinical phenotype, the preliminary diagnosis was HPS-5 subtype (Figure 5).
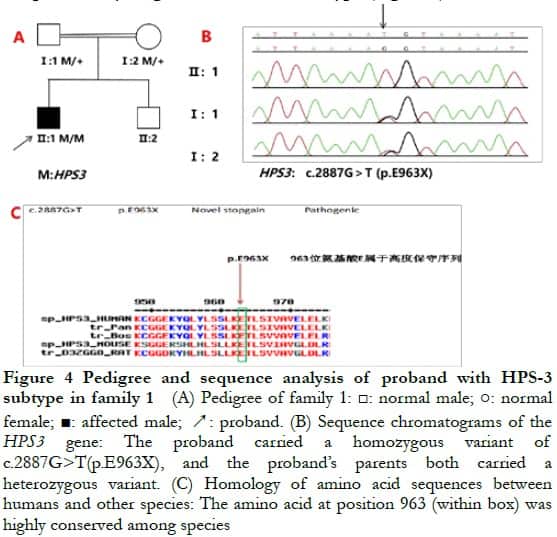
The variants identified from the two families were not included in HGMD and were new. The proband of family 1 carried the HPS3 gene c.2887G>T mutation leading to the early termination of peptide chain synthesis, and most of the proteins had loss of activity or normal function (PVS1). The conservatism analysis of amino acid showed that the amino acid sequence 963 of the HPS3 gene translation was highly conservative (Figure 4), and the changed amino acid led to the changes in protein structure and function and caused the disease. The mutation and disease were co-separated in the family (PP1_Supporting). The variation was not reported in the database of 1000 people and the database of East Asian population (ExAC_EAS) (PM2_Supporting). Therefore, it was predicted using LRT and Variant Taster software that the variant was a harmful variant with pathogenicity. According to the ACMG guidelines 5, the mutation was determined to be pathogenic.
The proband of family 2 carried the HPS5 gene complex heterozygous variation c.2952-2A>C splice mutation and chromosome 11 heterozygous deletion (3 144-bp deletion, located at chr11: 18,302,108-18,305,251, exon 22, PM2_Supporting). The c. 2952-2A>C mutation was located in the classical shear region, which destroyed the “GT-AG” shear structure, inferring that it might cause shear abnormality and affect protein coding (PVS1). The two variants were not reported in the 1000 population database and the East Asian population database (ExAC_EAS) (PM2_Supporting). The mutation and disease were co-separated in the family (PP1_Supporting). According to the ACMG guidelines, the mutation was determined to be pathogenic.
3 Discussion
The HPS3 gene (NCBI RefSeq: NM_032383) is located in 3q24 and contains 17 exons. The encoded HPS3 protein contains 1,004 amino acids. A total of 39 HPS3 gene variants have been reported in HGMD, including frameshift, duplication, nonsense, and missense variants 6. The HPS5 gene (NCBI RefSeq: NM_181507) is located at 11p15.1, including 23 exons, encoding the HPS5 protein containing 1129 amino acids. A total of 37 HPS5 gene variants have been reported in HGMD, most of which are frameshift and missense variants 7-8. The HPS-5 subgroup is relatively rare. At present, only two cases have been reported in China, and four variants of the HPS5 gene have been found 9-10. Three new variants were identified in the two families with HPS in this study. Family 1 had homozygous nonsense variation (parental consanguineous marriage), and family 2 had a compound heterozygous variation with splicing site and loss of heterozygosity. The aforementioned three new variants of HGMD were not included. The frequency in the normal population database was 0, the software prediction results showed that they were harmful variants, and the ACMG genetic variation classification standards and guidelines rated them as pathogenic variants.
HPS is a rare heterologous recessive genetic disease characterized by the abnormality of lysosomes and lysosomal-related organelles. HPS is characterized by eyelid albinism and bleeding tendency, accompanied by complications of various systems and organs of the body. The degree of hypopigmentation varies in different HPS subtypes. For example, the hair and skin colors of patients with HPS-1 can gradually deepen with age, and the hair and skin color of patients with the HPS-3 subtype can be normal or show mild hypopigmentation. In this study, the proband of family 1 with the HPS-3 subtype showed no obvious hypopigmentation in hair, eyebrows, and skin color. The patient complained that the hair color was brownish yellow when he was young, and gradually deepened with age, but the skin color did not change significantly. Whether the pigmentation of patients with the HPS-1 subtype will also occur in patients with the HPS-3 subtype with age needs further research to confirm. Tsilou et al10 reported that patients with HPS-3 had fewer ocular clinical manifestations compared with those with HPS-1. The proband of family 1 in this study had mild ocular and systemic clinical manifestations, but no severe visual impairment or fatal pulmonary fibrosis. The patients with HPS often have mild-to-moderate red and green vision abnormalities; however, the probands with the HPS-3 subtype had a normal color vision in this study. Jard ó n et al1 reported that, among the 64 patients with HPS, the BCVA of patients with the HPS-3 subtype was better than that of patients with the HPS-1 subtype. Exotropia was more common in patients with the HPS-3 subtype, while esotropia was more common in those with the HPS-1 subtype. The proband of pedigree 1 in this study showed exotropia. Bleeding tendency is one of the common phenotypes of patients with HPS. Most of these phenotypes are manifested as subcutaneous ecchymosis or skin bruises. Severe bleeding may occur after tooth extraction, surgery, or major trauma. Female patients may have excessive menstruation or postpartum bleeding. Therefore, patients with HPS need to avoid trauma and take appropriate hemostasis measures before surgery to prevent possible major bleeding11. The two probands in this study had a bleeding tendency, which was caused by the deficiency or large reduction of platelet dense granules leading to the defect of platelet storage pool. Platelet dense granules are lysosome-related organelles rich in H+, Ca2+, Zn2+, and so forth. After the platelets are stimulated and activated, platelet dense particles rapidly release their contents, such as adenosine diphosphate, to strengthen platelet aggregation reaction and also cause platelet second-phase aggregation reaction, thus promoting various physiological reactions such as coagulation and thrombosis. Recently, Yuan et al12 screened a zinc ion transporter, transmembrane protein163 (TMEM163), located in platelet dense granules. The absence of TMEM163 in mice leads to the deficiency of Zn2+ storage and biogenesis in platelet dense granules, and TMEM163 is significantly reduced in BLOC-1-, BLOC-2-, AP-3-deficient mice and patients with HPS, indicating that these complexes might participate in the transport of TMEM163 to platelet dense granules. The lack of these complexes or TMEM163 itself leads to the defect of platelet dense particles, resulting in coagulation dysfunction. This discovery revealed the key molecular mechanism of the defect of platelet dense particles, platelet storage pool disease, and platelet dense particle deficiency in HPS. In the present study, we observed the platelets of probands from the two families with HPS using an electron microscope. The findings revealed that the number of platelet dense particles significantly reduced, thus providing a clear basis for diagnosis. In addition, pulmonary fibrosis in patients with HPS showed many clinical, radiological, and histological characteristics of idiopathic pulmonary fibrosis, and the onset age was young. The first peak of incidence occurred at the age of 20–25 years. High resolution CT is the gold standard for diagnosing pulmonary fibrosis2,13. In this study, the proband with HPS-3 subtype failed to undergo high-resolution CT examination, and pulmonary CT examination showed no active lesions in both lungs. We will continue to follow up with the patient, closely observe the disease progression, and prevent complications. The proteins encoded by different HPS genes belong to different complexes and have different mechanisms of action. Different subtypes affecting the same BLOC or AP-3 often have similar clinical phenotypes. Di Pietro 14 and others also confirmed that the pathogenesis of HPS-3, HPS-5, and HPS-6 subtypes had a common biological basis. For example, the proband of family 2 in this study was the HPS-5 subtype, which had more clinical phenotypes similar to those of the HPS-3 subtype, such as horizontal nystagmus, exotropia, fundus pigment loss, and macular fovea dysplasia. Pulmonary CT showed that the number of normal, platelet dense particles significantly reduced, and blood coagulation function was basically normal. Meanwhile, some differences were also noted. For example, the color of the hair and eyebrows of the proband in family 2 had hypopigmentation compared with that in family 1; although the iris pigment was normal, the vision damage was serious. The clinical phenotypes of patients with different HPS subtypes have obvious heterogeneity, such as patients with HPS-3 and HPS-5 subtypes whose main characteristics are ocular albinism (OA) or OCA, accompanied by systemic complications, such as bleeding tendency and granulomatous colitis15. So far, nothing has been reported on HPS-3 and HPS-5 pulmonary fibrosis at home and abroad. Pulmonary fibrosis is a common complication in patients with HPS-1 and HPS-4 subtypes, which mainly shows progressive irreversible fibrosis of pulmonary parenchyma and pulmonary interstitium, and the patient may eventually die of respiratory failure. The common complication in patients with the HPS-2 subtype is a repeated infection caused by neutropenia. The typical clinical manifestation is chronic agranulocytosis. In addition, the severity of complications of each subtype belonging to different complexes is also significantly different: patients with HPS-3, HPS-5, and HPS-6 subtypes had mild symptoms, those with HPS-7, HPS-8, and HPS-9 subtypes showed severe symptoms, and those with HPS-1 and HPS-4 subtypes exhibited moderate symptoms 15.
Genetic testing is an important technical means to diagnose HPS. Using gene diagnosis technology to identify HPS subtypes helps clinicians reduce the occurrence of misdiagnosis and actively intervene in potential complications in patients. In this study, the proband of family 1 was diagnosed with the HPS-3 subtype by clinical examination and gene testing. With complete hemostasis measures, the proband of family 1 successfully underwent binocular strabismus correction surgery. The proband of family 2 was previously misdiagnosed with OCA. The sequencing in the whole-exon group revealed that the proband of family 2 had an HPS-5 subtype.
To sum up, family 1 and family 2 in this study had HPS-3 and HPS-5 subtypes, respectively, and a certain genotype and phenotype correspondence existed between the two types. The whole-exome sequencing technology could quickly and accurately screen candidate genes of HPS and identify clinical subtypes, which is conducive to early diagnosis of HPS and accurate intervention of potential complications by clinicians. For patients with HPS, systematic research using a large sample size is still lacking. The clinical phenotypes of each subtype are highly heterogeneous, and unified objective quantitative evaluation criteria are still lacking. For example, the judgment of the degree of pigment deficiency in fundus, hair, and skin is subjective, and the hair color of some subtypes of patients changes with age. The OCT examination to assess macular development was difficult because some patients were too young to cooperate or lack the ability of keep an eye on a target. Some cases are easily misdiagnosed as OA or OCA. Therefore, how to improve the relevant examination of HPS clinical phenotype, formulate objective quantitative evaluation criteria, and improve the diagnostic rate through gene detection are important directions of HPS research. It is suggested that patients with OA and OCA should be routinely screened for HPS genes to assess the risk of bleeding and the conditions of various organs and systems in the body and to sequence the whole-exon group as far as possible. Moreover, attention should be paid to the differential diagnosis with HPS, so as to avoid missed diagnosis or misdiagnosis of HPS.
Conflicts of interest None declared
Author contribution Yang Shangying: Subjects enrollment, article writing and revising; Cheng Wanyu:data colletion and analysis; Zhang Yan: electron microscopic examination and imaging data collection; Sheng Xunlun: Study conception, article revision
Acknowledgements Special thanks go to Professor Li Yang from Beijing Tongren Hospital and Professor Yuan Yefeng Beijing Children’s Hospital, Capital Medical University for their electron microscopic examination methods and operation skills
References
[1] Jardón J, Izquierdo NJ, Renta JY, et al. Ocular findings in patients with the Hermansky-Pudlak syndrome (types 1 and 3)[J]. Ophthalmic Genet, 2016, 37(1):89-94. DOI: 10.3109/13816810.2014.907920.
[2] El-Chemaly S, Young LR. Hermansky-Pudlak syndrome[J]. Clin Chest Med, 2016, 37(3):505-511. DOI: 10.1016/j.ccm.2016.04.012.
[3] Liu T, Yuan Y, Bai D, et al. Genetic variants and mutational spectrum of Chinese Hermansky-Pudlak syndrome patients[J]. Pigment Cell Melanoma Res, 2021, 34(1):111-121. DOI: 10.1111/pcmr.12916.
[4] Wei AH, Li W. Hermansky-Pudlak syndrome: pigmentary and non-pigmentary defects and their pathogenesis[J]. Pigment Cell Melanoma Res, 2013, 26(2):176-192. DOI: 10.1111/pcmr.12051.
[5] Richards S, Aziz N, Bale S,et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med, 2015, 17(5):405-24.DOI: 10.1038/gim.2015.30.
[6] Saito S, Tanaka R, Sasaki T, et al. Subclinical hypopigmentation of the skin and hair in a Japanese patient with Hermansky-Pudlak syndrome type 3[J/OL]. J Dermatol, 2020, 47(1):e18-e20[2022-04-05]. https://pubmed.ncbi.nlm.nih.gov/31621111/. DOI: 10.1111/1346- 8138.15118.
[7] Botero JP, Chen D, Majerus JA, et al. Hermansky-Pudlak syndrome subtype 5 (HPS-5) novel mutation in a 65 year-old with oculocutaneous hypopigmentation and mild bleeding diathesis: the importance of recognizing a subtle phenotype[J]. Platelets, 2018, 29(1):91-94. DOI: 10.1080/09537104.2017.1361019.
[8] Wei A, Yuan Y, Bai D, et al. NGS-based 100-gene panel of hypopigmentation identifies mutations in Chinese Hermansky-Pudlak syndrome patients[J]. Pigment Cell Melanoma Res, 2016, 29(6):702-706. DOI: 10.1111/pcmr.12534.
[9] Wei A, Yuan Y, Qi Z, et al. Instability of BLOC-2 and BLOC-3 in Chinese patients with Hermansky-Pudlak syndrome[J]. Pigment Cell Melanoma Res, 2019, 32(3):373-380. DOI: 10.1111/pcmr.12748.
[10] Tsilou ET, Rubin BI, Reed GF, et al. Milder ocular findings in Hermansky-Pudlak syndrome type 3 compared with Hermansky-Pudlak syndrome type 1[J]. Ophthalmology, 2004, 111(8):1599-1603. DOI: 10.1016/j.ophtha.2003.12.058.
[11] Power B, Ferreira CR, Chen D, et al. Hermansky-Pudlak syndrome and oculocutaneous albinism in Chinese children with pigmentation defects and easy bruising[J/OL]. Orphanet J Rare Dis, 2019, 14(1):52[2022-04-05]. https://pubmed.ncbi.nlm.nih.gov/30791930/. DOI: 10.1186/s13023-019-1023-7.
[12] Yuan Y, Liu T, Huang X, et al. A zinc transporter, transmembrane protein 163, is critical for the biogenesis of platelet dense granules[J]. Blood, 2021, 137(13):1804-1817. DOI: 10.1182/blood.2020007389.
[13] Lecchi A, La Marca S, Femia EA, et al. Novel variant in HPS3 gene in a patient with Hermansky Pudlak syndrome (HPS) type 3[J]. Platelets, 2020, 31(7):960-963. DOI: 10.1080/09537104.2019.1704716.
[14] Di Pietro SM, Falcón-Pérez JM, Dell’Angelica EC. Characterization of BLOC-2, a complex containing the Hermansky-Pudlak syndrome proteins HPS3, HPS5 and HPS6[J]. Traffic, 2004, 5(4):276-283. DOI: 10.1111/j.1600-0854.2004.0171.x.
[15] Liu T, Wei AH. Advances in Hermansky-Pudlak syndrome[J]. Dermatol Bull, 2020, 37(1):53-59.