·Clinical Research·
Identification of a COL2A1 mutation in a Chinese family with Stickler syndrome type 1 via whole exome sequencing
Deng Fang, Cao Yingjie, Xie Lijing, Chen Shaowan, Xiao Xiaoqiang, Zhang Mingzhi
Joint Shantou International Eye Center of Shantou University & The Chinese University of Hong Kong, Shantou 515041, China
Corresponding author: Xiao Xiaoqiang, Email: lijingyu198001980@163.com
[Abstract] [View PDF in English] [View PDF in Chinese] [Read Full Text]
Objective To identify genetic mutations associated with Stickler syndrome type 1 in a Chinese family.
Methods A pedigree investigation was conducted of the members of a Chinese family that included some individuals with Stickler syndrome type 1 recruited from the Shantou International Eye Center in June 2012. Medical histories were collected and clinical examinations were conducted, which included visual acuity, intraocular pressure, slit lamp microscopy, and fundus autofluorescence. The diagnoses were made by experienced clinicians. Total genomic DNA was extracted from peripheral blood samples (5 mL) collected from five affected and four healthy family members. The potential variant of the father of the proband (subject III-5) was screened by whole exome sequencing and stepwise bioinformatics analyses. Segregation and mutation conformation of the variant were verified by Sanger sequencing. The pathogenicity of the variant was predicted by SIFT, Polyphen2, and MutationTaster. Conservation and three-dimensional structure of the amino acid mutation were determined by multiple sequence alignment and tools available from the UniProt website.
Results Autosomal dominant inherence was identified in 39 members of the family, spanning four generations, which included 15 affected and 24 phenotypically normal individuals. The proband (subject IV-4) had retinal detachment of the right eye, binocular strabismus, and high myopia. Subject III-5 had high myopia of the right eye and cataract and atrophy of the left eye. Subject Ⅳ-9 had high myopia of both eyes. A heterozygous variation, c.1693C>T (p.Arg565Cys), within exon 26 of COL2A1 was found in the affected individuals, but not phenotypically normal individuals, demonstrating co-separation. The variant was predicted as deleterious by SIFT, Polyphen2, and MutationTaster. The amino acid residue at position 565, which was highly conserved among the human, mouse, rat, bovine, and Xenopus laevis sequences, exhibited an arginine to cysteine substitution in triple helix repeat region Gly-X-Y, thereby altering protein function.
Conclusions The COL2A1 variant c.1693C>T (p.Arg565Cys) was identified as the cause of Stickler syndrome type 1 in this family. This is the first report of this variant in China.
[Key words] Stickler syndrome, type 1; Pedigree; Genetic testing; Whole exome sequencing; Retinal detachment; High myopia; COL2A1 gene
Fund program: This study was financially supported by the Top-notch Talent Project of Guangdong Sailing Plan (grant no. 0142) and the Medical College Clinical Medicine High-level Key Discipline Construction Project of China
DOI: 10.3760/cma.j.cn115989-20200612-00425
Stickler syndrome is a genetic disorder of the connective tissues caused by mutations to collagen-encoding genes via autosomal dominant inheritance (COL2A1, COL11A1, or COL11A2) or autosomal recessive inheritance (COL9A1, COL9A2, or COL9A3)1-2. Collagens are important constitutes of the extracellular matrix of the chondrocyte, eye, and inner ear, and necessary for cartilage formation and growth, and maintenance of healthy joints, vision, and hearing3. Dysregulation of collagen biogenesis can affect the structure of eyes, ears, joints, and facial structures, resulting in defects of the ocular tissues (high myopia, vitreous degeneration, cataract, peripheral retinal degeneration, rhegmatogenous retinal detachment), sensorineural hearing loss, and facial deformities (saddle nose, micrognathia, cleft palate)4-5. Most cases of Stickler syndrome are due to COL2A1 mutations6-8. In light of the wide spectrum of hereditary disorders associated with collagen proteins, clinical manifestations are varied, complex, and difficult to diagnosis. Moreover, effective treatments are lacking for such diseases, especially rhegmatogenous retinal detachment. Therefore, the identification of pathogenic variants of Stickler syndrome will help to elucidate the molecular pathogenesis and contribute to disease diagnosis and personized medicine to enhance the quality of life of patients. Whole exome sequencing (WES) is widely used to identify genetic mutations associated with inheritable diseases. Here, WES was employed to identify a pathogenic variant of COL2A1 in members of a Chinese family with Stickler syndrome.
1 Materials and methods
1.1 General data
The study cohort included 39 members of a Chinese family (15 with Stickler syndrome and 24 phenotypically normal) spanning four generations and 200 healthy controls, which included older individuals with cataracts. Subject recruitment began in June 2021. Detailed clinic data were collected, which included information about deceased family members provided by relatives. The study protocol was approved by the Ethics Committee of the Joint Shantou International Eye Center (Shantou, Guangdong province, China) (approval no. EC20110310(2)-P02) and conducted in accordance with the ethical principles for medical research involving human subjects described in the Declaration of Helsinki. Prior to inclusion in this study, written informed consent was obtained from all subjects.
1.2 Methods
1.2.1 Clinic examinations Clinical examinations of all study subjects included visual acuity testing, slit-lamp examinations, and confocal laser scanning of the posterior segment of the eye (Heidelberg retina tomograph II).
1.2.2 Blood sample collection and DNA extraction Peripheral blood samples (5 mL) were collected from five affected individuals and four heathy controls in this family and 200 healthy individuals as controls recruited from the Joint Shantou International Eye Center. Genomic DNA was extracted from whole blood samples using the TIANamp Blood DNA Kit (Tiangen Biotech (Beijing) Co., Ltd., Beijing, China) in accordance with the manufacturer’s instructions and quantified with a NanoDrop™ 1000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The DNA samples were immediately used for WES or stored at −20°C.
1.2.3 WES and screening of candidate pathogenic variants WES of the genomic DNA of subject III-5 was conducted by a commercial sequencing service (Annoroad Gene Technology Co., Ltd., Beijing, China). In brief, WES of 2 µg DNA was performed using the SureSelect Human All Exon Kit V5 (Agilent Technologies, Inc., Santa Clara, CA, USA). Paired-end sequencing of each sample was conducted with the HiSeq 2500 platform (PE100; Illumina, Inc., San Diego, CA, USA) with read lengths of 100 bp and average coverage depth of at least 100×. The raw WES data were used to screen for mutations. Single-nucleotide polymorphisms and insertion/deletions were detected with SAMtools (http://www.htslib.org/) by mapping the divergent reads against the University of California, Santa Cruz human reference genome hg19 with the Burrows–Wheeler Aligner (https://sourceforge.net/projects/bio-bwa/files/). To screen for candidate variants, high-frequency variants (minor allele frequency > 0.01) in the 1000 Genomes project (https://www.internationalgenome.org/) and Genome Aggregation Database (https://gnomad.broadinstitute.org/) were initially excluded, followed by all intergenic variants, intronic variants, and synonymous mutations. Then, the data were checked for mutations to all genes associated with Stickler syndrome. Remaining mutations were analyzed using the protein structure prediction programs Polymorphism Phenotyping v2 (Polyphen-2; http://genetics.bwh.harvard.edu/pph2/) and Sorting Intolerant From Tolerant (SIFT; https://sift.bii.a-star.edu.sg/). Polyphen-2 uses sequence- and structure-based predictive algorithms and generates a unique scale of reported scores of the effects of identified mutations on protein function. The SIFT tool is mainly used to identify the positions of conserved amino acids among different species to predict the effects of missense changes on protein structure.
1.2.4 Sanger sequencing and co-segregation analysis Amplification by polymerase chain reaction (PCR) and Sanger sequencing were conducted to confirm the identified candidate variants. PCR was performed using a real-time PCR detection system (Bio-Rad Laboratories, Inc., Hercules, CA, USA) with a primer pair specific for COL2A1 (forward: 5’-CCC TTG GCT TCA GAC CCT-3’; reverse: 5’-CCC CTG TCA CAA TTC TCA AAA TT-3’) designed with Primer3 software (https://primer3.org/). The PCR products were purified with an E.Z.N.A.® Cycle Pure Kit (Omega Bio-Tek, Inc., Norcross, GA, USA) and sequenced by Guangzhou IGE Biotechnology Ltd. (Guangzhou, China). The mutations detected among the five affected and four healthy family members were subjected to co-segregation analysis and excluded from the sequences of the 200 healthy controls by Sanger sequencing
1.2.5 Conserved amino acids and three-dimensional protein structures Alignments of multiple protein sequences and prediction of three-dimensional protein structures were performed with tools available from the UniProt website (https://www.uniprot.org/).
2 Results
2.1 Clinic manifestation
The cohort of 39 individuals from the same Chinese family spanning four generations included 15 individuals with Stickler syndrome and 24 otherwise healthy individuals (Figure 1). The proband (subject IV-4) was a 7-year-old boy with high myopia, retinal detachment, heterotropia of the right eye, and blindness of the left eye. Intraocular pressure of the right and left eye was 10 and 6 mmHg, respectively. Following retinal detachment surgery combined with silicone oil tamponade, unaided vision of the right eye recovered to 0.1 (1 year later) with intraocular pressure of 33.1 mmHg (considered secondary glaucoma) (Figure 2). Subject III-5 had high myopia and cataract of the right eye and atrophy of the left eye. Subject III-14 had staphyloma and sensory esotropia of the right eye and atrophy of the left eye. Subject IV-9 had high myopia of both eyes (right eye: −17.00 OD; left eye: −18.00 OD). Other affected individuals within this family showed similar clinic symptoms of Stickler syndrome.

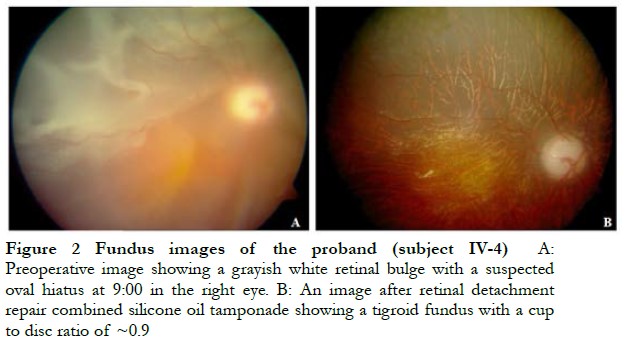
2.2 WES and identification of candidate variants
Step-wise screening of the WES data identified a heterozygous mutation to the COL2A1 gene (c.1693C>T: p.565Arg>Cys), which was predicted as deleterious with SIFT, Polyphen2, and MutationTaster (https://www.mutationtaster.org/).
2.3 Sanger sequencing and co-segregation assay
Analysis of the variant revealed that all of the tested affected individuals carried the mutation, but not the healthy family members or 200 controls. As shown in Figure 3, the Sanger sequencing results confirmed that the variant co-segregated with the disease phenotype, suggesting the variant was a pathogenic mutation.

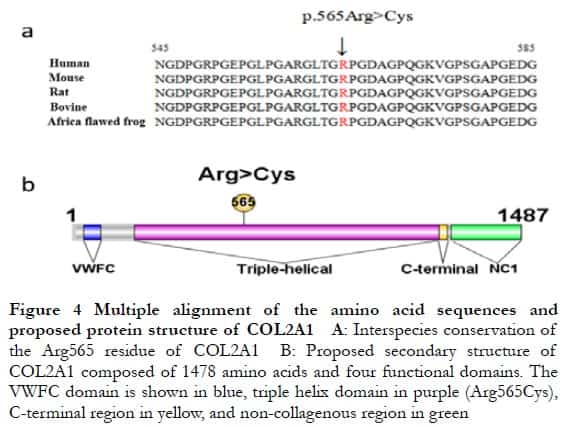
2.4 Location of the mutated amino acid of the COL2A1 protein
Alignment of the partial COL2A1 protein sequences containing the variant of the human, mouse, rat, bovine, and African clawed frog showed that the mutated arginine residue at position 565 (Arg565) is highly conserved (Figure 4A). The Arg565 residue located within the triple-helical domain of COL2A1 mutated to cysteine (Cys) and might have compromised the stability of the Gly-X-Y sequence (Figure 4B), resulting in possible loss some important biological functions linked to the pathogenesis of Stickler syndrome.
3 Discussion
The common clinic manifestations of Stickler syndrome include high myopia, vitreous abnormalities, cataract, peripheral retinal degeneration, and rhegmatogenous retinal detachment. Other clinic symptoms include micrognathia (alone or as part of the Pierre Robin sequence), sensorineural hearing loss, early onset osteoarthritis, spondyloepiphyseal dysplasia, and cleft palate. These clinic manifestations tend to be more obvious in early childhood. Notably, high myopia is not present in all patients with Stickler syndrome and tends to be stable rather than progressive 5. In this study, the diagnosis of Stickler syndrome was based on clinic examinations conducted at the Joint Shantou International Eye Center and other hospitals. The age of onset of high myopia was 7 years for subject IV-4 and 5 years for subject IV-9. Moreover, two of the affected family members had retinal detachment. However, some of the clinic data obtained from the other hospitals lacked information about hearing loss, skeletal/joint malformations, and craniofacial abnormalities. Nonetheless, the lack of clinic data did not affect the diagnoses or conclusions of this study that a deleterious variant of the COL2A1 gene was associated with the pathogenesis of type 1 Stickler syndrome.
Mutations to COL2A1, which encodes type II collagen, COL11A1 and COL11A2, which encode type XI collagen, and COL9A1 and COL9A2, which encode type IX collagen, have been associated with type I, II, III, IV, and V Stickler syndrome, respectively. COL2A1 and COL11A1 are mainly expressed in the vitreous. Mutations to COL2A1 usually result in a congenital membranous vitreous anomaly. In contrast, mutations to COL11A1 result in a different vitreous phenotype where the gel that fills the posterior chamber forms beaded bundles with irregular diameters, probably reflecting the role of type XI collagen in the regulation of collagen fibrillogenesis 5-6. COL11A2 is not expressed in the eye, thus a mutation leads to Stickler syndrome without eye involvement. Mutations in COL9A1 result in rapid aging of the vitreous due to progressive liquefaction and degeneration, leading to formation of an optical vitreous cavity. Mutation to COL9A2 results in similar phenotypes as types 1 and 2. COL2A1 contains 54 exons, but pathogenic variants have not been identified in exons 3, 5, 22, 24, 31, 37, or 54 9-31. Some pathogenic variants are located within introns 10, 14-15, 29, 32. Exon 2, which encodes the VWFC domain 9, 15, 17, 28, exon 23 9, 10, 15, 28, 30-31, exon 42 9, 10, 15, and exon 51 9, 10, 14-15, 23 carry most variants, suggesting that these exons are mutation hotspots. Other exons contain fewer variants, but are more evenly distributed. Of the 166 reported variants of COL2A1, 155 (93.4%) are located within the triple-helical domain of COL2A1. Among the amino acid residues, the frequency of mutation is greater for the glycine and Arg residues. Most of the frame shift mutations cause vitreous-related symptoms. The COL2A1 variant found in the patient cohort of this study was previously reported in two sporadic cases. Sporadic case MS12 exhibited symptoms similar to those of subject IV-4, which included retinal detachment, high myopia, and heterotopia, while the symptoms of sporadic case MS16 were similar to those of subject III-5, which included high myopia, retinal detachment, and cataract. The Arg565Cys mutation, which was first reported in a Chinese patient, has been linked to the clinical manifestations of Stickler syndrome 10.
In conclusion, the results of this study found that the COL2A1 variant c.1693C>T (p.Arg565Cys) was linked to the clinical manifestations of type 1 Stickler syndrome.
Conflict of interest None declared.
Author contributions DF perform the experiments and analyzed the data. CYJ and CSW analyzed the data. XLJ collected the clinical and diagnostic data. XXQ conceived and designed the study, arranged funding, performed the experiments, analyzed and interpreted the data, and revised the article.
References
[1] Stickler GB, Belau PG, Farrell FJ, et al. Hereditary progressive arthro-ophthalmopathy[J]. Mayo Clin Proc, 1965, 40:433–455.
[2] Francomano CA, Liberfarb RM, Hirose T, et al. The Stickler syndrome: evidence for close linkage to the structural gene for type II collagen[J]. Genomics, 1987, 1(4):293-296. DOI: 10.1016/0888-7543(87)90027-9.
[3] Spranger J, Winterpacht A, Zabel B. The type II collagenopathies: a spectrum of chondrodysplasias[J]. Eur J Pediatr, 1994, 153(2):56-65. DOI: 10.1007/BF01959208.
[4] Matsushita I, Nagata T, Hayashi T, et al. Foveal hypoplasia in patients with Stickler syndrome[J]. Ophthalmology, 2017, 124(6):896-902. DOI: 10.1016/j.ophtha.2017.01.046.
[5] Snead MP, Yates JR. Clinical and molecular genetics of Stickler syndrome[J]. J Med Genet, 1999, 36(5):353-359.
[6] Richards AJ, McNinch A, Martin H, et al. Stickler syndrome and the vitreous phenotype: mutations in COL2A1 and COL11A1[J/OL]. Hum Mutat, 2010, 31(6):E1461-1471[2022-02-15]. https://pubmed.ncbi.nlm.nih.gov/20513134/. DOI: 10.1002/humu.21257.
[7] Van Camp G, Snoeckx RL, Hilgert N, et al. A new autosomal recessive form of Stickler syndrome is caused by a mutation in the COL9A1 gene[J]. Am J Hum Genet, 2006, 79(3):449-457. DOI: 10.1086/506478.
[8] Baker S, Booth C, Fillman C, et al. A loss of function mutation in the COL9A2 gene causes autosomal recessive Stickler syndrome[J]. Am J Med Genet A, 2011, 155A(7):1668-1672. DOI: 10.1002/ajmg.a.34071.
[9] Richards AJ, Baguley DM, Yates JR, et al. Variation in the vitreous phenotype of Stickler syndrome can be caused by different amino acid substitutions in the X position of the type II collagen Gly-X-Y triple helix[J]. Am J Hum Genet, 2000, 67(5):1083-1094. DOI: 10.1016/S0002-9297(07)62938-3.
[10] Kondo H, Matsushita I, Nagata T, et al. Novel mutations in the COL2A1 gene in Japanese patients with Stickler syndrome[J/OL]. Hum Genome Var, 2016, 3:16018[2022-02-15]. https://pubmed.ncbi.nlm.nih.gov/27408751/. DOI: 10.1038/hgv.2016.18.
[11] Higuchi Y, Hasegawa K, Yamashita M, et al. A novel mutation in the COL2A1 gene in a patient with Stickler syndrome type 1: a case report and review of the literature[J/OL]. J Med Case Rep, 2017, 11(1):237[2022-02-16]. https://pubmed.ncbi.nlm.nih.gov/28841907/. DOI: 10.1186/s13256-017-1396-y.
[12] Huang X, Lin Y, Chen C, et al. Targeted next‑generation sequencing identifies two novel COL2A1 gene mutations in Stickler syndrome with bilateral retinal detachment[J]. Int J Mol Med, 2018, 42(4):1819-1826. DOI: 10.3892/ijmm.2018.3752.
[13] Gerth-Kahlert C, Grisanti S, Berger E, et al. Bilateral vitreous hemorrhage in a newborn with Stickler syndrome associated with a novel COL2A1 mutation[J]. J AAPOS, 2011, 15(3):311-313. DOI: 10.1016/j.jaapos.2011.03.008.
[14] Wubben TJ, Branham KH, Besirli CG, et al. Retinal detachment and infantile-onset glaucoma in Stickler syndrome associated with known and novel COL2A1 mutations[J]. Ophthalmic Genet, 2018, 39(5):615-618. DOI: 10.1080/13816810.2018.1509355.
[15] Hoornaert KP, Vereecke I, Dewinter C, et al. Stickler syndrome caused by COL2A1 mutations: genotype-phenotype correlation in a series of 100 patients[J]. Eur J Hum Genet, 2010, 18(8):872-880. DOI: 10.1038/ejhg.2010.23.
[16] Wang X, Jia X, Xiao X, et al. Mutation survey and genotype-phenotype analysis of COL2A1 and COL11A1 genes in 16 Chinese patients with Stickler syndrome[J]. Mol Vis, 2016, 22:697-704.
[17] Yoshida S, Yamaji Y, Kuwahara R, et al. Novel mutation in exon 2 of COL2A1 gene in Japanese family with Stickler syndrome type I[J]. Eye (Lond), 2006, 20(6):743-745. DOI: 10.1038/sj.eye.6702001.
[18] Brown DM, Vandenburgh K, Kimura AE, et al. Novel frameshift mutations in the procollagen 2 gene (COL2A1) associated with Stickler syndrome (hereditary arthro-ophthalmopathy)[J]. Hum Mol Genet, 1995, 4(1):141-142. DOI: 10.1093/hmg/4.1.141.
[19] Yoon JM, Jang MA, Ki CS, et al. Two likely pathogenic variants of COL2A1 in unrelated Korean patients with ocular-only variants of Stickler syndrome: the first molecular diagnosis in Korea[J]. Ann Lab Med, 2016, 36(2):166-169. DOI: 10.3343/alm.2016.36.2.166.
[20] Guo L, Elcioglu NH, Wang Z, et al. Novel and recurrent COL11A1 and COL2A1 mutations in the Marshall-Stickler syndrome spectrum[J/OL]. Hum Genome Var, 2017, 4:17040[2022-02-10]. https://pubmed.ncbi.nlm.nih.gov/28983407/. DOI: 10.1038/hgv.2017.40.
[21] Yaguchi H, Ikeda T, Osada H, et al. Identification of the COL2A1 mutation in patients with type I Stickler syndrome using RNA from freshly isolated peripheral white blood cells[J]. Genet Test Mol Biomarkers, 2011, 15(4):231-237. DOI: 10.1089/gtmb.2010.0138.
[22] Baijens LW, De Leenheer EM, Weekamp HH, et al. Stickler syndrome type I and stapes ankylosis[J]. Int J Pediatr Otorhinolaryngol, 2004, 68(12):1573-1580. DOI: 10.1016/j.ijporl.2004.07.015.
[23] Zechi-Ceide RM, Jesus Oliveira NA, Guion-Almeida ML, et al. Clinical evaluation and COL2A1 gene analysis in 21 Brazilian families with Stickler syndrome: identification of novel mutations, further genotype/phenotype correlation, and its implications for the diagnosis[J]. Eur J Med Genet, 2008, 51(3):183-196. DOI: 10.1016/j.ejmg.2007.12.008.
[24] Vu CD, Brown J Jr, Körkkö J, et al. Posterior chorioretinal atrophy and vitreous phenotype in a family with Stickler syndrome from a mutation in the COL2A1 gene[J]. Ophthalmology, 2003, 110(1):70-77. DOI: 10.1016/s0161-6420(02)01446-x.
[25] Williams CJ, Ganguly A, Considine E, et al. A-2–>G transition at the 3′ acceptor splice site of IVS17 characterizes the COL2A1 gene mutation in the original Stickler syndrome kindred[J]. Am J Med Genet, 1996, 63(3):461-467. DOI: 10.1002/(SICI)1096-8628(19960614)63:3<461::AID-AJMG9>3.0.CO;2-U.
[26] Liu X, Dong H, Gong Y, et al. A novel missense mutation of COL2A1 gene in a large family with stickler syndrome type I[J]. J Cell Mol Med, 2022, 26(5):1530-1539. DOI: 10.1111/jcmm.17187.
[27] McAlinden A, Majava M, Bishop PN, et al. Missense and nonsense mutations in the alternatively-spliced exon 2 of COL2A1 cause the ocular variant of Stickler syndrome[J]. Hum Mutat, 2008, 29(1):83-90. DOI: 10.1002/humu.20603.
[28] Wang DD, Gao FJ, Hu FY, et al. Next-generation sequencing-aided precise diagnosis of Stickler syndrome type I[J/OL]. Acta Ophthalmol, 2020, 98(4):e440-e446[2022-02-12]. https://pubmed.ncbi.nlm.nih.gov/31736238/. DOI: 10.1111/aos.14302.
[29] Čopíková J, Paděrová J, Románková V, et al. Expanding the phenotype spectrum associated with pathogenic variants in the COL2A1 and COL11A1 genes[J]. Ann Hum Genet, 2020, 84(5):380-392. DOI: 10.1111/ahg.12386.
[30] Chu FC, Hii LY, Hung TH, et al. A novel de novo mutation in COL2A1 gene associated with fetal skeletal dysplasia[J]. Taiwan J Obstet Gynecol, 2021, 60(2):359-362. DOI: 10.1016/j.tjog.2021.01.017.
[31] Lauritsen KF, Lildballe DL, Coucke PJ, et al. A mild form of Stickler syndrome type II caused by mosaicism of COL11A1[J]. Eur J Med Genet, 2017, 60(5):275-278. DOI: 10.1016/j.ejmg.2017.03.005.
[32] Sun W, Xiao X, Li S, et al. A novel deep intronic COL2A1 mutation in a family with early-onset high myopia/ocular-only Stickler syndrome[J]. Ophthalmic Physiol Opt, 2020, 40(3):281-288. DOI: 10.1111/opo.12682.