·Clinical research·
Pathogenic mutation screening in a family with Axenfeld-Rieger Syndrome by whole exome sequencing
Wang Qi, Liu Xinna, Shao Zhengbo, Yuan Huiping
Department of Ophthalmology, the Second Affiliated Hospital of Harbin Medical Universiiy, The Key Laboratory of Myocardial Ischemia, Harbin Medical University, Ministry Educatton, Future Medical Laboratory, the Second Affiilated Hospital of Harbin Medical Universiy, Harbbn 150086, Chiia
Corresponding author: Yuan Huiiing, Email: yuanhp2013@126.com
[Abstract] [View PDF in English] [View PDF in Chinese] [Read Full Text]
Objective This study was aimed at identifying disease-causing genetic variation in a Chinese family with Axenfeld-Rieger syndrome (ARS) through the analysis of clinical symptoms and hereditary information.
Methods The method of pedigree investigation was used. A Chinese family with ARS including 15 pedigree members over three generations was recruited at the Second Affiliated Hospital of Harbin Medical University in 2018. The family included three patients with ARS. The family history and clinical data were collected. Ophthalmic and general examinations were performed in all family members included in the study. DNA and RNA were extracted from collected peripheral venous blood samples (2–5 ml) from each family member. Whole exome sequencing was used to screen the variations in the proband. Suspected variations screened through searching of population databases and bioinformatics analysis were verified by Sanger sequencing and real-time quantitative PCR. Conservation analysis and deleteriousness prediction of suspected variations were conducted. The pathogenicity of candidate rare variations was evaluated according to American College of Medical Genetics and Genomics (ACMG) standards and guidelines. This study followed the Declaration of Helsinki. The study protocol was approved by the Ethics Committee of the Second Affiliated Hospital of Harbin Medical University (No. KY2019-231). Written informed consent was obtained from each participant or custodian before entering the study cohort.
Results The three patients with ARS had typical clinical features of ARS in the eyes, teeth and umbilicus, and carried the same heterozygous variant, c.525delC•(p.Asp175Glufs•), in the PITX2 gene; this variant was not found in other family members, thereby indicating co-segregation. The relative expression of PITX2 mRNA was 0.672 ± 0.063 in the patients with ARS, which was significantly lower than the 1.015 ± 0.179 observed in the healthy controls (t = 8. 847, P < 0.001). This variant was not recorded in the dbSNP, 1000G, gnomeAD, ExAC, Korea 1K, or Exome Variant Server (EVS) databases, and it was denoted deleterious by MutationTaster. The affected conservative amino acid sequences were found in nine species. The variant was determined to be pathogenic according to the ACMG standards and guidelines.
Conclusions The c.525delC (p.Asp175Glufs•) mutation of the PITX2 gene was pathogenic in the pedigree. This study provides the first report of this mutation in a Chinese family with ARS.
[Keywords] Axenfeld-Rieger syndrome; Pedigree; Genetic testing; Whole exome sequencing; PITX2 gene; Frameshift mutation
Fund program: National Natural Science Foundation of China (82070956); Applied Technology Research and Development
Program of Heilongjiang Provincial Science and Technology Department (GA20C008); Postgraduate Research & Practice Innovation Program of Harbin Medical University (YJSSJCX2019-45HYD)
DOI: 10.3760/cma.j.cn115989-20200802-00555
Axenfeld-Rieger syndrome (ARS) is a group of rare developmental genetic diseases that involve the iris and angle, with an incidence of
1/100,000 to 1/50,000 in newborns [1]. In addition ARS shows no significant ethnic or sex differences and is mostly autosomal dominant. Sporadic ARS cases have occasionally been reported. ARS is characterized by a posterior corneal embryonic ring, anterior iris adhesions, and iris dysplasia in the eyes; glaucoma is present in approximately 50% of cases. Patients with ARS may also have jaw dysplasia and dental, umbilical, and cardiac developmental abnormalities. However, the clinical manifestations in patients vary greatly according to the location and severity of the developmental abnormalities involved. Iris abnormalities can manifest as mild stromal thinning, textural abnormalities, pupil displacement, iris fissures (i.e., multiple pupils), and pupil deformation resulting from stromal defects. In addition, patients with slight ocular abnormalities and no systemic developmental defects are easily misdiagnosed or missed; therefore, clear genetic diagnosis not only is conducive to guiding prenatal and postnatal care and understanding the pathogenesis, but also can provide a reference for clinical diagnosis. Moreover, to improve the prognosis of patients, physicians must pay attention to other systemic developmental abnormalities throughout the body and provide early intervention. The PITX2 and FOXC1 genes, which encode transcription factors, are currently known ARS pathogenic genes, which are detected in only 40% of patients with ARS [1]. Because ARS is genetically heterogeneous, genetic diagnosis is not possible for all patients through traditional ARS pathogenic mutation screening methods, e.g., Sanger sequencing of known pathogenic genes. Whole exome sequencing (WES) can rapidly and efficiently detect exonic region variants in the genome that affect protein structure, and can rapidly target rare suspected pathogenic variants in combination with bioinformatics methods. In this study, WES combined with Sanger sequencing validation was used to detect gene mutations and analyze mutational pathogenicity in patients with ARS, with the aim of clarifying ARS diagnosis to provide accurate clinical diagnosis, treatment, and genetic counseling for patients and their pedigree members, and providing a genetic basis for studying the mechanisms of ARS.
1 Materials and methods
1.1 General data
The pedigree study method was used to analyze a Chinese Han family with anterior segment dysplasia who visited the Department of Ophthalmology of the Second Affiliated Hospital of Harbin Medical University in 2018. The medical history and family history were collected in detail. The proband was 10 years old at the time of the visit, and the pedigree comprised a total of 15 individuals in three generations, including three patients with ARS. The study was conducted in accordance with the Declaration of Helsinki, and the study protocol was approved by the Medical Ethics Committee of the 2nd Affiliated Hospital of Harbin Medical University (approval No.: KY2019-231). Pedigree members who participated in this study or their guardians were informed of the objective of this study and voluntarily signed the informed consent form.
1.2 Methods
1.2.1 Ophthalmic and general examination All members involved in the study underwent professional ophthalmic examination. The best corrected visual acuity was examined with a standard logarithmic hyperopia chart, and the anterior segment was examined with a slit lamp microscope (SL-1E, Topcon, Japan). Intraocular pressure was measured in healthy individuals with a non-contact tonometer (CT-80, Topkon, Japan) and in patients with ARS with a rebound tonometer (TA01i, iCare, Finland). Anterior segment photography was performed with a slit-lamp image system (YZ5T, Suzhou 66 Vision Technology Co., Ltd.) to record the anterior segment and angle. Moreover, the angle abnormalities were observed and recorded with a digital wide-field imaging system for children (RetCam, CREE, USA), and the retinal and choroidal findings were recorded through color fundus photography with a digital fundus camera (CR-2, Canon, Japan). A general examination of the whole body was also performed, and if necessary, a specialist examination was performed.
1.2.2 Genomic DNA extraction A total of 2–5 mL of peripheral venous blood was collected from pedigree members who agreed to genetic analysis. Samples were stored in EDTA anticoagulant tubes at −80 °C for future use. Genomic DNA was extracted according to the instructions of the Peripheral Blood Genomic DNA Extraction Kit [DP304, Tiangen Biotech (Beijing) Co., Ltd.].
1.2.3 Exome sequencing A Qubit instrument was used to measure the DNA sample concentration of the proband, and agarose gel electrophoresis was used to verify the sample quality. The sample was sent for WES. Exon capture was performed with an Agilent SureSelect Human Whole Exon V6 Kit (Agilent, USA), and sequencing was performed on the NovaSeq 6000 platform (Illumina, USA) after library construction.
1.2.4 Raw data processing and annotation After data with adapters and low quality data obtained by sequencing were filtered out, the valid data obtained were compared with the reference genome (GRCh37/hg19) in BWA software. After the comparison results were sequenced with SAMtools, and the repeats were labeled with Picard, GATK software was used to detect single nucleotide polymorphism (SNP) and insertion/deletion (InDel) variants. SnpEff software was used to structurally annotate the mutation sites and filter out variants that had been included in dbSNP with allele frequencies > 0.05. The result files were re-annotated with the wANNOVAR tool [2].
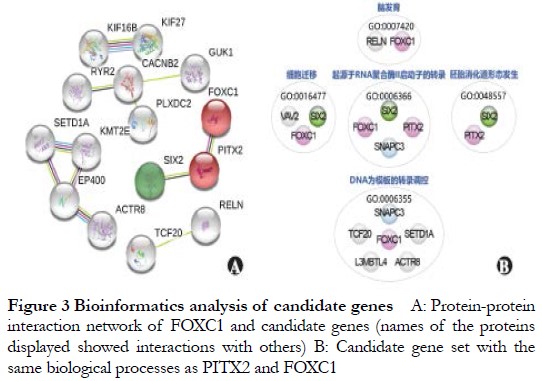
1.2.5 Screening for suspected pathogenic mutations in combination with population databases and bioinformatic assessments The SNP and InDel variants obtained after annotation were screened according to the following steps: (1) splice variants and exonic region variants were retained, and non-coding variants and synonymous variants were excluded; (2) variants with global or Asian allele frequencies < 0.00001 or unlisted variants in the 1000 Genomes Project (1000G), Exome Aggregation Consortium (ExAC), and Genome Aggregation Database (gnomAD) Genomic and Exome Sequencing Database were retained; (3) the data on WES variants in individuals with no blood relationship within the group and in this pedigree were compared to exclude common variants present in more than two individuals simultaneously; (4) retained SIFT, PolyPhen-2, MutaionTaster, or CADD (Phred > 20) were predicted as deleterious genetic variation; (5) reported ARS-associated genes were searched; and (6) other related mutations were screened through protein interaction network analysis of the suspected mutant genes through the Search Tool Retrieval of Interacting Genes (STRING) database[3-4] for PITX2 and FOXC1 related proteins, and Gene Ontology (GO) analysis of suspected genes through the Database for Annotation, Visualization and Integrated Discovery (DAVID) online tool [5] to identify genes with the same biological processes annotation entries as PITX2 and FOXC1.
1.2.6 Sanger sequencing co-isolation validation and pathogenicity assessment Specific amplification primers were designed for suspected pathogenic mutations, and after the target fragment was amplified with the PCR reagent Gold Mix (TSE101, Tsingke Biotechnology Co., Ltd.) on an S1000 ™ Thermo Cycler PCR instrument (Bio-Rad, USA), false positive results were excluded by Sanger sequencing validation, and co-segregation of the mutation between genotypes and clinical phenotypes in normal pedigree members and patients with ARS was verified. Primer synthesis and Sanger sequencing were performed by the Harbin Branch of Tsingke Biotechnology Co., Ltd., and Chromas software was used to compare and analyze the sequencing results. The conservation analysis of amino acids in nine animal species (human, chimpanzee, Rhesus monkey, dog, cow, mouse, rat, chicken and zebrafish) was performed with UGENE software [2,6-7]. The Korea Genome Project (Korea 1K) and Exome Variant Server (EVS) databases were used to verify the allele frequency of the mutation in the population. Candidate rare variants were assessed according to the criteria and guidelines of the American College of Medical Genetics and Genomics (ACMG) for classification of genetic variants [8].
1.2.7 Detection of PITX2 mRNA expression in lymphocytes of healthy individuals and participants by real-time PCR mRNA was extracted from peripheral blood lymphocytes of three patients and two healthy individuals in the pedigree, and one healthy individual unrelated to the pedigree, according to the instructions of the ultrapure RNA extraction kit (CW0581S, Beijing Cowin Biotech Co., Ltd), then reverse transcribed into cDNA with a reverse transcription kit (Transcriptor First Strand cDNA Synthesis Kit, Roche, Switzerland) and detected with real-time PCR on a C1000 ™ Thermo Cycler fluorescence PCR instrument (Bio-Rad, USA) with NCSYB GREEN qPCR Master Mix (NC-BM601, Harbin Nachuan Biotech Co., Ltd.). PITX2 had a forward primer sequence of 5´-GACATGTACCCAGGCTA TTCC-3′ and reverse primer sequence of 5´-GGGTTGACGTTCATAG AGTTGA-3, and GAPDH was used as an internal reference, with a forward primer sequence of 5´-GTCTCCTCTGACTTCAACAGCG-3´ and reverse primer sequence of 5-ACCACCCTGTTGCTGTAGCCAA-3′. The reaction conditions were as follows: pre-denaturation at 95 ℃ for 15 min, denaturation at 95 ℃ for 10 s, and annealing and extension at 56 ℃ for 30 s for 39 cycles. The relative expression of PITX2 mRNA was calculated with the 2-ΔΔCt method.
1.3 Statistical methods
GraphPad Prism 8.0.2 statistical software was used for statistical analysis. Measurement data confirmed to be normally distributed by the Shapiro-Wilk test are expressed as x̅±s. The relative expression of target gene mRNA between patients and healthy controls was compared with independent samples t-tests. A p-value < 0.05 indicated statistically significant differences.
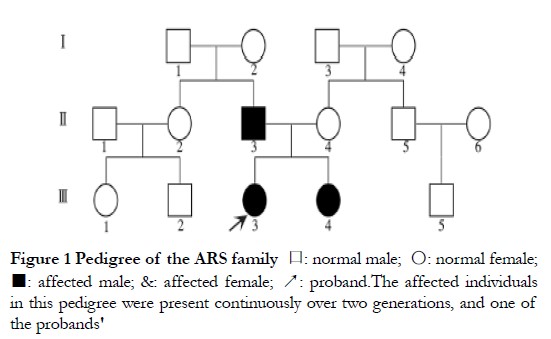
2 Results
2.1 Clinical characteristics of pedigree members
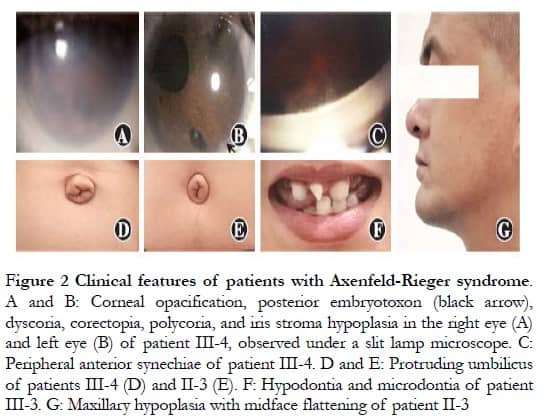
Parents were affected, in agreement with the known autosomal dominant inheritance characteristics of ARS; however, the possibility of X-chromosome dominant inheritance was not excluded (Figure 1). Patients II-3, III-3, and III-4 presented with varying degrees of corneal opacity, posterior corneal embryonic rings, iris dysplasia, pupil displacement, and anterior synechia, with elevated intraocular pressure in both eyes (Figures 2A–C). Proband III-3 had grayish opacities in the cornea of the left eye, a faintly visible iris and bullae in the corneal epithelium of the left eye, scattered scar structures in the subepithelium, and nerve fiber loss, according to laser scanning confocal microscopy. All three patients had a flat midface, tooth defects, and canine teeth and umbilical skin protrusion (Figures 2D–G). No ocular or systemic abnormalities were observed in patients I-2 or III-4.
2.2 WES sequencing analysis
The coverage of the WES target region of probands 1–3 was 97.74%, and the percentage of bases with a depth not less than 30× was 93.24%, with an average depth of coverage of more than 100× for all chromosomes. Non-rare variants in public and internal databases were excluded from the sequencing results, and 121 mutations with high impact on protein sequences were retained. According to the predictions of bioinformatics software, two InDel variants, one splicing mutation, and three nonsense mutations were evaluated as known deleterious or potentially deleterious by MutationTaster; 35 missense mutations were evaluated as deleterious by CADD and SIFT, and deleterious by at least one of MutationTaster and Polyphen-2, including the InDel variant of the ARS pathogenic gene PITX2. The genes involved in the above 41 mutations were applied as candidates for further bioinformatics analysis of known ARS pathogenic genes, and genes with protein interaction scores greater than 0.4 (moderate confidence) were selected for STRING analysis. SIX2 interacted with PITX2 (Figure 3A), and DAVID annotation revealed that eight genes that had similar functions to PITX2 or FOXC1 were located under GO biological processes annotation entries, such as cell migration, transcriptional regulation with DNA as a template, and transcription originating from RNA polymerase and promoter (Figure 3B).
2.3 Validation and analysis of suspected pathogenic mutation
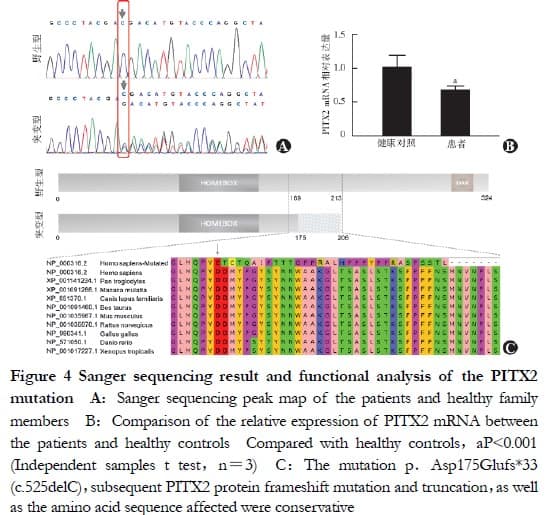
The heterozygous mutation c.525delc in the ARS pathogenic gene PITX2 (NM_000325.6) was involved in the above candidate gene mutation, which was found to be carried by II-3, III-3, and III-4 in the pedigree validation, i.e., all family members clinically diagnosed with ARS in the pedigree. However, I and I-2, and n-4, with a normal phenotype, did not carry this mutation, in agreement with pedigree co-segregation (Figure 4A). The relative expression of PITX2 mRNA in the peripheral blood lymphocytes of the three patients was 0.672 ± 0.063, a value significantly lower than that of the healthy controls (1.015 ± 0.179) (t = 8.847, P < 0.001, Figure 4B). The variant was not included in the dbSNP, 1000G, gnomeAD, ExAC, Korea1K, or EVS databases, and was indicated as deleterious by MutationTaster. A frame-shift mutation, p.Asp175 glufs. (Np _ 000316.2), was predicted at the protein level, with a truncated sequence length from 324 to 206 amino acids and the deletion of the OAR domain, a conserved structural domain of the PITX2 protein. The affected amino acid sequence was conserved in nine animal species (Figure 4C). According to ACMG genetic variation classification criteria and guidelines, the PITX2 gene mutation c.525delC/p.Asp175Glufs in this pedigree be accordant to 1 PS evidence, three PM evidence, and 2 PP evidence, thus demonstrating that the mutation was pathogenic.
3 Discussion
In this study, the frame-shift mutation c.525delC (p.Asp175Glufs •) in the PITX2 gene was reported for the first time in a Chinese family, thus demonstrating that PITX2 pathogenic mutation is associated with ARS. Beyond ARS, this gene mutation in the eye is associated with iris dysplasia, iridoctral angle dysplasia syndrome, Peters’ abnormality, and corneal dermoid rings [9-11]. Mutations in the PITX2 pathogenic gene are also observed in patients with dental dysplasia and with no ocular manifestations, congenital heart disease, or atrial fibrillation [12-15]. Phenotypic diversity caused by PITX2 mutations may be attributable to the following. (1) Because dental and cardiac abnormalities may also occur in patients with ARS, incomplete penetrance of the phenotype may result in clinical differences. (2) Differences exist in the genetic background of each patient; for example, the presence of SNPs in other genes in the body may alter the susceptibility of patients to certain disease phenotypes, thus causing phenotypic differences. In the reported ARS pedigrees, the ocular phenotype of patients was almost completely penetrant, and all patients in this study had ocular anomalies and similar dental and umbilical developmental defects, with no differences in the sites of disease involvement, and differences only in severity. Because patients with the same PITX2 gene mutation have different sites of disease involvement in the individual ARS pedigree [16], we speculated that the phenotypic heterogeneity found in the same pedigree might have been associated with genetic background differences between patients. In the ARS genetic study, data on other gene variations in the ARS pedigree should be obtained in addition to the pathogenic genes of the disease, and differences in the genetic background of the pedigree should be analyzed to reveal the cause of phenotypic heterogeneity in patients with ARS and the mechanism of ITX2-induced dysplasia.
Currently, the high-throughput sequencing technologies used for parallel sequencing of many gene sequences are mainly WES, whole genome sequencing, and target gene sequencing (TGS), among which TGS and WES are widely applied for variant screening of monogenic genetic diseases [17]. TGS, also known as panel sequencing, can detect only mutations in the target gene fragment, and the panel is selected on the basis of the patient’s clinical diagnosis [18]. WES, which can compensate for the above drawbacks, is suitable for diseases in which causative genes have not been fully identified. In this study, in addition to the known ARS causative gene PITX2 variants, some rare mutations in other genes that may affect protein structure were identified with WES technology. The genes involved exhibited similar gene functions to the known pathogenic genes PITX2 or FOXC1: the protein encoded by the SIX2 gene interacts with the PITX2 protein. However, the pathogenicity of these mutations is currently unknown, and further pedigree-based genetic analysis and mutation function studies are required to investigate the correlations between these candidate genes and ARS pathogenesis and phenotypic differences.
Like other eye-associated genetic diseases, ARS has allele heterogeneity in addition to locus heterogeneity [17], and ARS-associated PITX2 gene mutations are mainly in the form of truncated mutations (nonsense mutations or frame-shift mutations) and missense mutations in important functional domains. More than 100 variants have been reported to date, including approximately 45 missense and nonsense mutations, approximately 23 base insertions or deletions, and approximately 4 mutations in the splice region. No significant correlation has been found between the form and location of the PITX2 gene mutation and the severity of patient phenotype. However, systemic dysplasia, such as craniofacial, dental, and navel dysplasia, is more common in patients with this gene mutation than in patients with ARS with the FOXC1 gene mutation [1]. The c.525delC (p.Asp175Glufs •) frame-shift mutation identified in this study altered the amino acid sequence conserved in vertebrates and resulted in the deletion of the OAR domain at and within the PITX2 protein terminus. In addition, the OAR structure at the PITX2 protein terminus and its flanking 39 amino acids were associated with the DNA binding and transcriptional activation of PITX2 protein [19]. Thus, the c.525 delC heterozygous mutation might alter the function of the PITX2 protein as a gain-of-function or loss-of-function mutation; cause disease by inhibiting the function of wild-type PITX2 protein, owing to the dominant negative mutation effect of the translated mutant protein; or cause ARS because of haploinsufficiency of the PIG gene resulting from frame-shift mutation-mediated mRNA degradation. According to the detection of PITX2 mRNA expression, the expression of PITX2 mRNA was diminished in patients with ARS with this mutation. Moreover, on the basis of bioinformatics analysis, the protein sequence of this mutation was predicted to be truncated by 118 amino acids (approximately 36%) after transcription and translation; therefore, the mRNA transcribed by the mutant DNA chain was likely to be degraded. However, further investigation is required to determine whether the mutant gene is eventually translated into a functional protein and what its effects at the organism level might be.
In conclusion, the pathogenic heterozygous mutation c.525delC (p.Asp175Glufs) in the PITX2 gene was identified in an ARS pedigree in northern China through WES technology in combination with Sanger sequencing, thus providing the first report of an ARS pedigree in China. In this study, the clinical and genetic diagnosis of patients in this pedigree was clarified, thus aiding in diagnosis and treatment in line with disease characteristics, expanding the PIX2 gene mutation spectrum to a Chinese pedigree, and highlighting the roles of PITX2 in the formation of ARS and the occurrence of development-associated genetic diseases.
Conflict of interests None declared.
Author contribution Wang Qi: Study conception and implementation, data collection, statistical analysis and manuscript writing; Liu Xinna: Study implementation, data collection, statistical analysis and manuscript revision; Shao Zhengbo: Study guidance and manuscript revision; Yuan Huiping: Subject collection, study guidance and manuscript revision
References
[1] Seifi M, Walter MA. Axenfeld-Rieger syndrome[J]. Clin Genet, 2018, 93(6):1123-1130. DOI: 10.1111/cge.13148.
[2] Chang X, Wang K. wANNOVAR: annotating genetic variants for personal genomes via the web[J]. J Med Genet, 2012, 49(7):433-436. DOI: 10.1136/jmedgenet-2012-100918.
[3] Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets[J/OL]. Nucleic Acids Res, 2019, 47(D1):D607-D613[2022-03-01]. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6323986/. DOI: 10.1093/nar/gky1131.
[4] Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible[J/OL]. Nucleic Acids Res, 2017, 45(D1):D362-D368[2022-03-03]. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5210637/. DOI: 10.1093/nar/gkw937.
[5] Huang DW, Sherman BT, Tan Q, et al. DAVID bioinformatics resources: expanded annotation database and novel algorithms to better extract biology from large gene lists[J/OL]. Nucleic Acids Res, 2007, 35(Web Server issue): W169-175[2022-03-23]. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC 1933169/. DOI: 10.1093/nar/gkm415.
[6] Rose R, Golosova O, Sukhomlinov D, et al. Flexible design of multiple metagenomics classification pipelines with UGENE[J]. Bioinformatics, 2019, 35(11):1963-1965. DOI: 10.1093/bioinformatics/bty901.
[7] Okonechnikov K, Golosova O, Fursov M, et al. Unipro UGENE: a unified bioinformatics toolkit[J]. Bioinformatics, 2012, 28(8):1166-1167. DOI: 10.1093/bioinformatics/bts091.
[8] Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology[J]. Genet Med, 2015, 17(5):405-424. DOI: 10.1038/gim.2015.30.
[9] Xia K, Wu L, Liu X, et al. Mutation in PITX2 is associated with ring dermoid of the cornea[J/OL]. J Med Genet, 2004, 41(12):e129[2022-03-10]. https://pubmed.ncbi.nlm.nih.gov/15591271/. DOI: 10.1136/jmg.2004.022434.
[10] Kozlowski K, Walter MA. Variation in residual PITX2 activity underlies the phenotypic spectrum of anterior segment developmental disorders[J]. Hum Mol Genet, 2000, 9(14):2131-2139. DOI: 10.1093/hmg/9.14.2131.
[11] Doward W, Perveen R, Lloyd IC, et al. A mutation in the RIEG1 gene associated with Peters’ anomaly[J]. J Med Genet, 1999, 36(2):152-155.
[12] Intarak N, Theerapanon T, Ittiwut C, et al. A novel PITX2 mutation in non-syndromic orodental anomalies[J]. Oral Dis, 2018, 24(4):611-618. DOI: 10.1111/odi.12804.
[13] Sun YM, Wang J, Qiu XB, et al. PITX2 loss-of-function mutation contributes to tetralogy of Fallot[J]. Gene, 2016, 577(2):258-264. DOI: 10.1016/j.gene.2015.12.001.
[14] Wei D, Gong XH, Qiu G, et al. Novel PITX2c loss-of-function mutations associated with complex congenital heart disease[J]. Int J Mol Med, 2014, 33(5):1201-1208. DOI: 10.3892/ijmm.2014.1689.
[15] Qiu XB, Xu YJ, Li RG, et al. PITX2C loss-of-function mutations responsible for idiopathic atrial fibrillation[J]. Clinics (Sao Paulo), 2014, 69(1):15-22. DOI: 10.6061/clinics/2014(01)03.
[16] Perveen R, Lloyd IC, Clayton-Smith J, et al. Phenotypic variability and asymmetry of Rieger syndrome associated with PITX2 mutations[J]. Invest Ophthalmol Vis Sci, 2000, 41(9):2456-2460. DOI: 10.1007/s004170000130.
[17] Chinese Genetic Eye Diseases Diagnosis Workgroup, Chinese Genetic Eye Diseases Alliance. Experts consensus on genetic eye diseases diagnosis [J]. Chin J Exp Ophthalmol, 2018, 36(7):481-488. DOI: 10.3760/cma.j.issn.2095-0160.2018.07.001.
[18] Li Y. Clinical phenotype assessment is very important in mutation analysis for patients with hereditary eye disease[J]. Chin J Exp Ophthalmol, 2017, 35(8):673-676. DOI: 10.3760/cma.j.issn.2095-0160.2017.08.001.
[19] Amendt BA, Semina EV, Alward WL. Rieger syndrome: a clinical, molecular, and biochemical analysis[J]. Cell Mol Life Sci, 2000, 57(11):1652-1666. DOI: 10.1007/pl00000647.